Causes of Friedreich's ataxia
A person can get this pathology only if both of his parents are carriers of the mutating gene. The mutation occurs in the long arm of the ninth chromosome, which provokes disturbances in the synthesis of the frataxin protein from mitochondria, which, in turn, act as “cellular energy stations.”
Iron accumulates in mitochondria and is oxidized. Oxygen transport occurs in the body. When iron synthesis is disrupted, its amount in mitochondria increases sharply and significantly (about ten times). In this case, cellular iron remains within normal limits, and the level of cytosolic iron decreases.
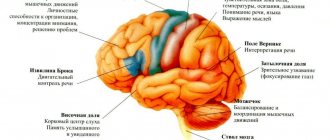
Such processes activate genes that encode fragments responsible for iron transport—ferroxidases and permeases. The intracellular iron balance is further disrupted. As a result of a high concentration of iron in the cell, radicals are activated, which have a damaging effect and destroy the cell from the inside. The most vulnerable cells are neurons (especially in the posterior and lateral columns of the spinal cord, in the spinocerebellar tracts, and peripheral nerve fibers).
Considering the degree of gene mutation, “classical” forms of the disease and atypical, so to speak lighter versions, benign syndromes are distinguished.
Friedreich's hereditary ataxia is the most common ataxia.
1.General information
Ataxia (ancient Greek “disorder”) is a neurological pathological phenomenon consisting of incoordination and chaotic motor skills: complex neuromuscular relationships are disrupted, as a result of which the patient’s movements become unfocused and unproductive. Ataxia, expressed to one degree or another, accompanies many diseases of the central nervous system (CNS), for example, atrophic and tumor disorders, as well as some psychopathological disorders, but a group of ataxias is distinguished as independent diseases.
In particular, more than two dozen variants of hereditary spinocerebellar (i.e., caused by combined damage to the spinal cord and cerebellum) ataxia, or SCA, are known, which is a genetically determined degenerative process in these structures of the central nervous system.
Friedreich's familial ataxia (named after the physician who gave the first clinical description of the disease in 1860) is the most common of the hereditary neurodegenerative diseases with a predominance of ataxia in the clinical picture. It occurs, according to various sources and with some regional dependence, in 3-7 people for every hundred thousand population; passive carriage of a defective gene is much more common (about 1%). The probability of starting the process does not depend on gender.
A must read! Help with treatment and hospitalization!
Symptoms of Friedreich's ataxia
The clinical picture develops more clearly between the ages of 10 and 20 years, although it is possible that symptoms of Friedreich’s ataxia may be detected at a later age. There is a hypothesis that the classic and atypical forms of this disease can be caused by various mutations of one or more genes. The first symptoms most often appear during the formation of the reproductive system.
The clinical picture is characterized by a combination of neurological and extraneural symptoms. Before the advent of DNA diagnostics, the clinical picture of the disease was described only in the classical form. Later, scientists came to the conclusion that the spectrum of the disease is much deeper and the prevalence is higher, so they began to identify erased and atypical forms of Friedreich's ataxia.
Among the neurological symptoms of Friedreich's ataxia are:
- A feeling of awkwardness and uncertainty when walking (appears as one of the very first symptoms), which intensifies if a person is in the dark. There is staggering, the person often stumbles, and unmotivated falls occur. There is instability in the Romberg position and the inability to repeat the knee-heel test. Over time, disorientation appears in the hands, legs quickly get tired, and handwriting may change. With outstretched arms, tremor is observed, and the finger-nose test is impossible (the patient constantly misses).
- Dysarthria (may not be observed in all cases), disorders of the speech apparatus.
- There is a disturbance or complete disappearance of tendon and periosteal reflexes (observed already in the early period of the clinical picture, and is an important diagnostic link).
- Inhibition of the Achilles and knee reflexes (sometimes appear long before the onset of other signs of the disease).
- Total areflexia (more often occurs in the advanced stage).
- Impaired joint-muscular and vibration sensitivity.
- Babinski's sign (extension of the big toe as a reaction to painful stimulation of the foot) is one of the earliest signs of the disease.
- Muscular hypotonia.
- Weakness of the legs and decreased muscle tone can give way to complete atrophy.
- Cerebellar and sensory ataxia.
- Over time, hand ataxia, amyotrophy, deep sensitivity disorder may occur, and motor functions decay, which ultimately leads to the impossibility of self-care.
- Nystagmus (tremor of the eyelid), atrophy of the optic and auditory nerves, mental weakness, in the absence of adequate treatment, dysfunction of the pelvic organs may be observed.
Extraneural symptoms include:
- Heart damage.
- Increasing hypertrophic or dilated cardiomyopathy (pain in the heart, rapid heartbeat, frequent shortness of breath, even with minor exertion, systolic heart murmur). Often it is cardiomyopathy as a concomitant disease that is the cause of death in Friedreich's ataxia.
Often, electrocartographic symptoms significantly precede the neurological signs of Friedreich's ataxia (sometimes by several years), so it can be very difficult to correctly diagnose this disease. Patients most often spend a long time registered with a cardiologist with a diagnosis of rheumatic heart disease.
Signs of this disease that are also important for diagnosis are skeletal deformities:
- severe scoliosis;
- Friedreich's foot (the arch of the foot is highly concave, the toes are hyperextended in the main phalanges and bent in the distal phalanges);
- kyphoscoliosis;
- the fingers of the upper and lower extremities are deformed.
Such signs, like cardiomyopathy, may appear long before neurological symptoms.
With Friedreich's ataxia, a disorder of the endocrine system is observed, which can manifest itself in the form of the following diseases:
- ovarian dysfunction;
- diabetes;
- infantilism;
- hypogonadism.
Very often, patients with Friedreich's ataxia have cataracts, so they are also considered part of the clinical picture of this disease.
Friedreich's ataxia is characterized by rapid progression and increasing symptoms. The duration of the disease is often no more than twenty years.
The pronounced clinical picture of atypical Friedreich's ataxia is observed later than in the classical form - approximately in the third to fifth decade of a person's life.
The course occurs in a more mild form than with classical ataxia and the outcome of the disease is more favorable:
- The patient retains the ability to self-care for a long time.
- There is no development of diabetes mellitus.
- There are no paresis, reflexes are preserved.
Such clinical cases are described under the name “late Friedreich's ataxia” or “Friedreich's ataxia with preserved reflexes.”
Friedreich's ataxia (AF)
- an autosomal recessive disease, i.e. sick children are born to a pair of parents who are both clinically healthy, but are carriers of a pathological gene. The disease affects neurons of the central and peripheral nervous system: the Gaulle bundles are predominantly affected, to a lesser extent the Burdach, Flexig, Gowers bundles, as well as the pyramidal tracts, dorsal roots, spinal ganglia and peripheral nerves, cells of the cerebellar cortex, basal ganglia, cerebral cortex , conducting pathways of the spinal cord. In other systems, no less important organ cells are affected by the disease, these are myocardial cells, β - cells of the islets of Langerhanz in the pancreas, cells of the retina and bone tissue. Causes progressive degeneration of the central and peripheral nervous system. Most patients are homozygous; their mRNA content is so low that sometimes it is not detected at all (unlike healthy individuals and carriers of the Friedreich's ataxia gene). AF occurs equally often in both men and women, does not occur in representatives of the Negroid race, and almost never occurs in native Asians. Named after the German physician Nikolaus Friedreich, who first described it in 1860. It is the most common of the hereditary ataxias: the incidence of Friedreich's ataxia is about 2-7 cases per 100 thousand people.
| Symptoms of Friedreich's ataxia most often appear in the first and second decades of life, occasionally in the third and fourth decades. There is uncertainty, staggering, stumbling when walking, frequent falls, handwriting is impaired due to tremor, dysarthria appears, weakness in the legs, and hearing is impaired. Tendon and periosteal reflexes (primarily Achilles and knee) disappear. Sometimes rheumatic heart disease may be an early symptom. Patients do not perform the heel-knee test, swaying appears in the Romberg position, which intensifies when closing the eyes, and sitting disorders. Babinski's sign. Often nystagmus. Deep sensitivity is gradually impaired, muscle atrophy increases, in the initial stages it is more pronounced in the lower extremities, and as the disease progresses, it also affects the upper extremities. Total areflexia is formed. The optic nerve atrophies, cataracts develop, which leads to blindness, the function of the pelvic organs is disrupted, and dementia develops. Endocrine disorders develop: diabetes mellitus, hypogonadism, infantilism, ovarian dysfunction. Cardiomyopathies. Skeletal deformities: curvature of the spine, kyphoscoliosis, “Friedreich’s foot” (high concave arch of the foot with hyperextension of the toes in the main phalanges and flexion in the distal phalanges), deformation of the fingers and toes, clubfoot. Course of the disease Steadily progressing, in the absence of adequate treatment, the duration of the disease usually does not exceed 20 years. The immediate cause of death may be heart and pulmonary failure, infectious complications. In rare cases, in the absence of diabetes and cardiac disorders, patients live up to 70-80 years. The prognosis is more favorable in women: 100% of women and only 63% of men live more than 20 years from the onset of the disease. |
Diagnostics:
Computed tomography of the brain, which remains the main diagnosis of ataxia in this disease, is ineffective, because detects changes only in late stages. It is possible to detect only a weak degree of cerebellar atrophy at an early stage and atrophy of the hemispheres, expansion of the stem cisterns, lateral ventricles and subarachnoid space of both hemispheres at later stages. Early diagnosis of Friedreich's ataxia is made using MRI, which makes it possible to detect spinal cord atrophy and a decrease in the transverse size of the spinal cord, especially increasing in the caudal direction at the advanced stage, and moderate atrophy of the pons, cerebellum and medulla oblongata. At the initial stage, an electrophysiological study is required; during such studies, the severity of damage to the sensitivity of the nerves of the limbs is established. The electroneuromyographic pattern characteristic of this disease is the absence or significant decrease in the amplitude of action potentials in the sensory nerves of the extremities, with a relatively small decrease in the speed of impulse conduction along the motor nerves. For a complete diagnosis, stress tests of glucose tolerance (to rule out diabetes mellitus) and x-ray examination of the spine are performed. The ECG shows rhythm disturbances, T-wave inversion, conduction changes; with echocardiography, conduction disturbances, up to complete blockade, and hypertrophy of the interventricular septum are especially often observed. In some cases, clinical and electrocardiographic symptoms of cardiac damage sometimes precede the appearance of neurological disorders by several years. Patients are observed for a long time by a cardiologist or local physician, most often with a diagnosis of rheumatic heart disease. To assess mitochondrial disorders using the cytochemical method, it seems most appropriate to determine the activity of a number of lymphocyte dehydrogenase enzymes: succinate dehydrogenase (SDH),
Differentiation of diagnosis:
Similar symptoms are characteristic of cerebellar tumors, hereditary metabolic diseases: Gm 1- and Gm 2-gangliosidosis and galactosialidosis (for differentiation, the activity of β-galactosidase and hexosaminidase A is studied), neurosyphilis, funicular myelosis, hereditary ataxia with vitamin E deficiency, Bassen's syndrome Konzweig, hereditary metabolic diseases, such as Krabbe disease (differential - study of the enzyme galactosylceramidase) and late variant of Niemann-Pick disease (for differentiation, the content of sphingomyelins in the cerebrospinal fluid is determined, sternal puncture is examined for the presence of “foamy” cells), Louis-Bar disease (or otherwise ataxia-telangiectasia: clinically it is distinguished by the presence of telangiectasia on the skin (excessive local dilatation of small vessels, mainly precapillaries and capillaries), the absence of skeletal abnormalities, frequent and severe respiratory tract infections, the absence or extremely low level of IgA, high levels of a- fetoprotein. MRI reveals hypoplasia of the cerebellum, most often of its vermis.), with multiple sclerosis (differential diagnosis usually does not cause difficulties, since symptoms such as tendon areflexia, muscle hypotonia, amyotrophies, extraneural manifestations, and also due to with the absence of remissions and focal changes in the density of brain matter in Friedreich's disease on CT and MR imaging). To differentiate the disease, DNA testing and medical genetic counseling, blood lipid profile testing, and blood smear analysis for the presence of vitamin E and acanthocyte deficiency are performed. DNA testing should be prescribed not only to the patient, but also to relatives to determine the heredity of the disease; this is necessary for the purposes of prevention and prescribing preventive therapy. During a molecular genetic examination of patients with clinically typical manifestations of FA for an increase in the GAA trinucleotide, the expansion of the allele is not detected in all. In this case, a point mutation or deletion in the FD gene on both chromosomes is possible. In these cases, it may be a phenocopy of AF, since triplet enlargement has been described in many patients with atypical ataxia and in patients with generalized chorea.
Treatment
Friedreich's ataxia does not lead to complete recovery, but timely prevention makes it possible to avoid the development of many symptoms and complications. To slow the progression of the disease, mitochondrial drugs, antioxidants and other drugs that reduce the accumulation of iron in mitochondria are prescribed. The general principle of treatment with these drugs is the combined administration of drugs that synergistically affect different levels of energy metabolism. It is recommended to simultaneously prescribe at least three drugs from the first three groups. (Drugs that increase the activity of the mitochondrial respiratory chain, cofactors of enzymatic reactions of energy metabolism, antioxidants). Antioxidants such as vitamins A and E are prescribed, as well as a synthetic substitute for coenzyme Q 10 - idebenone, which inhibits the neurodegenerative process and the development of hypertrophic cardiomyopathy. Usually, drugs are prescribed that improve myocardial metabolism: riboxin, cocarboxylase, preductal, etc. 5-hydroxyprofane is also prescribed, which gives good results, but requires further research. In general, treatment is symptomatic, aimed at symptoms such as diabetes mellitus and diseases of the cardiovascular system. General strengthening treatment (vitamins) is carried out, as well as drugs that affect tissue metabolism (piracetam, aminalon, acephen, cerebrolysin), treatment of which should be repeated periodically. Surgical correction of the feet and injection of botulinum toxin into spastic muscles are also performed. Physiotherapy and exercise therapy are procedures without which treatment of Friedreich's ataxia most often turns out to be ineffective. Constant exercise makes it possible to keep the body in good shape and eliminate painful sensations. Children with FA can stay active as long as possible through physical therapy and corrective exercise routines that focus on balance and muscle strength. Cardiomyopathy does not develop with this exercise program. Patients feel better when limiting carbohydrates in food to 10 g/kg, since their high consumption is a kind of “provocation” that increases the defect in energy metabolism. Patients require social adaptation, as many have to live in a state of complete helplessness. Loss of vision, the ability to move independently, and impaired coordination create psychological disorders that must be eliminated with the help of specialists and the support of loved ones.
Prevention of Friedreich's ataxia
— DNA testing at an early presymptomatic stage is of particular importance in order to prescribe preventive therapy. The patient's relatives are examined first.
The prevalence of autosomal recessive Friedreich's ataxia among the Yakut population of the PC (Yakutia) is 2.8 per 100 thousand population. The molecular genetic cause of AF in Yakuts is the expansion (GAA) of n-repeat in intron 1 of the FRDA gene. In the uluses of Vilyui and central Yakutia, isolated cases of autosomal recessive Friedreich's ataxia without accumulation in individual uluses have been recorded. An interesting fact is that the disease was registered in the Yakut ethnic group, which is a representative of the Asian race, among whom no cases of Friedreich's ataxia had previously been registered. This can be explained by the introduction of a European component into the Yakut gene pool, therefore, elucidating the reasons for the occurrence and spread of Friedreich's ataxia among the Yakuts requires further studying.
Literature:
- DNA diagnosis of Friedreich's ataxia with clinical genetic analysis. // V. V. Pugachev, S. N. Illarioshkin, L. V. Prokutsha, B. D. Markova, 0. V. Evgrafov, I. A. Ishnova-Smolenskaya // Collection “Molecular diagnosis of hereditary diseases and medical and genetic counseling. “- M - MOLIHKll - 1QS3.
- Hereditary ataxia and paraplegia // S.N. Illarioshkin. // “Medicine”, 2006.
- Hereditary diseases of the nervous system.// Yu.E. Veltishchev, P.A. Temin. // “Medicine”, 1998.
- Neurology of childhood // A.S. Petrukhin. // “Medicine”, 2004.
- Neurology. Handbook of a practical physician // D.R. Shtulman, O.S. Levin // MEDpress-inform Publishing House, 2005.
- Detection of a polymorphic marker in the region of the gene responsible for the occurrence of Friedreich's ataxia. // O. V. Evgrafov, V. V. Pugachev, L. B. Strelchenko. // 2nd All-Union Symposium “Theoretical and Applied Aspects of Molecular Biology”. Samarkand. - 1991.- theses house. — P. 68.
- Search and study of microsatellite repeats in the area of the putative localization of the FRDA gene // O.V. Evgrafov, V.V. Pugachev, A.V. Polyakov, L. B. Strelchenko // II Congress of VOGIS, Minsk, 1992, Abstracts of reports. part 1.p.25
CT scan
The lack of adequate treatment accelerates the progression of the disease and leads it to a severe stage. The main diagnostic method for all ataxias is computed tomography of the brain. But in this case, it is ineffective, since most changes in the brain with Friedreich's ataxia are detected only in the later stages. This is explained by the spinal localization of changes. Early stages of the disease are not visible on CT scans. In the later stages, only minor atrophies of the cerebellum and hemispheres, some expansion of the cerebral cisterns, lateral ventricles, and subarachnoid space can be detected.
Treatment of the disease
Adequate and regular treatment of Friedreich's ataxia makes it possible to stop the progression of the disease, avoid complications, and maintain the patient's ability to lead an active lifestyle for a long time.
As a rule, Friedreich's ataxia is treated by the simultaneous administration of metabolic drugs belonging to 3 different groups: cofactors of energetic enzyme reactions, stimulators of mitochondrial respiratory chain activity and antioxidants.
Additionally, for Friedreich's ataxia, medications are prescribed that improve metabolic processes in the heart muscle (cocarboxylase, riboxin, preductal, 5-hydroxyprofan, etc.), nootropics and neuroprotectors (aminalon, piracetam, acephen, cerebrolysin, encephabol), and multivitamins.
Massage and physical therapy are of great importance for patients with Friedreich's ataxia. Regular exercise therapy aimed at training coordination and muscle strength makes it possible to maintain motor activity and relieve emerging pain. Children with FA can stay active as long as possible through physical therapy and corrective exercise routines that focus on balance and muscle strength. Cardiomyopathy does not develop with this exercise program.
Physiotherapeutic procedures such as thermal procedures (ozokerite, paraffin) on the area of the forearms and shins, and electrical stimulation of muscles have a good effect.
Since Friedreich's ataxia is accompanied by a violation of energy metabolism, patients with this disease need to limit the intake of carbohydrates from food, the excess of which can provoke aggravation of metabolic disorders.
A person suffering from Friedreich's ataxia needs some surgical interventions (mainly for the spine and heart). Titanium screws and rods are inserted into the spine to help prevent or slow the progression of scoliosis. The goal of the operation is to preserve the patient's condition for as long as possible. In many cases, the patient develops heart disease. These diseases can be treated with medication.
In some cases, surgical correction of foot deformities and injection of botulinum toxin into spastic muscles are performed.
Patients need social adaptation, since many have to live in a state of complete helplessness. Loss of vision, the ability to move independently, and impaired coordination create psychological disorders that must be eliminated with the help of specialists and the support of loved ones.
To stop the progression of ataxia, assistive devices such as a cane, walker, or wheelchair are used. Rehabilitation measures include a number of physical and therapeutic activities.
MRI
MRI is prescribed, with the help of which it is possible to detect atrophy in the spinal cord in the early stages, and also examines the transverse dimensions of the spinal cord. With Friedreich's ataxia they are below normal. Moderate atrophy of the pons, cerebellum and medulla oblongata is also visible.
Using an electrophysiological study, the degree of damage to the sensitivity of the nerves of the limbs is determined. With Friedreich's ataxia, the amplitude of the action potentials of the sensitivity of the nerves of the limbs is significantly reduced or completely absent.
Symptoms
The primary symptom of Friedreich's syndrome is progressive limb ataxia during walking. Ataxia involves inadequate muscle coordination, resulting in unstable gait and poor control of fine limb movements.
Involvement of the muscles of the mouth and throat can lead to slurred speech and difficulty swallowing. Intelligence is usually not affected.
A lateral curvature of the spine (scoliosis) or foot abnormalities may develop.
A form of heart disease (cardiomyopathy) is present in more than half of people with the disorder.
Since there is no effective therapy, clinical signs progress, requiring people to use a wheelchair for mobility.
Differential diagnosis
For an objective diagnosis, the patient must undergo a consultation with several doctors: an endocrinologist, an ophthalmologist, an orthopedist, and a cardiologist.
Diagnosis of this genetic disease is a difficult process due to the difficulty of differentiating the disease from a number of other, almost identical, and often concomitant diseases:
- Hereditary ataxia due to a lack of vitamin E. For differentiation, the concentration of vitamin E in the blood is determined, the blood lipid profile is examined, and the presence of acanthocytosis is detected using a blood smear.
- Bassen-Kornzweig syndrome.
- Diseases associated with metabolic disorders that are inherited in an autosomal recessive manner. For example, Krabbe disease, Niemann-Pick disease.
- Multiple sclerosis.
Causes
The gene responsible for the disorder is FXN. It encodes frataxin, a protein essential for the proper functioning of mitochondria, which produce energy for cells. Because both copies of the gene are abnormal, they do not produce enough frataxin protein.
Find out more Signs, diagnosis and treatment of Rotor syndrome
Tissues that are particularly dependent on cellular energy production—nerve cells and cardiac cells—begin to degenerate.
In most affected individuals, the FXN gene contains a very specific type of error called a GAA trinucleotide extended repeat.
Each gene consists of four chemical units (nucleotides) called adenine (A), cytosine (C), guanine (G), thymine (T).
In most people with the disorder, both copies of the FXN gene contain abnormally long stretches of guanine-adenine-adenine repeat units (GAA trinucleotide repeat). While healthy people have fewer than 30 GAA repeats, people with FRDA get extended tracts of 100 to 1300 repeats on both copies of the FXN gene. Most contain >400 repeats.
Although this extended GAA repeat mutation is located in the "non-coding" region of FXN (intron 1), it results in gene freezing and reduced ability to produce the frataxin protein. The severity of the disease and variability of symptoms are proportional to the length of the extended remutation.
For example, shorter repeats (<400 GAA) are associated with later age of onset, slower progression of clinical signs, and absent or mild cardiomyopathy.
Parents of children with Friedreich syndrome who have one copy of the repeat GAA mutation and one normal FXN gene do not show any signs of the disease.
Most people with the disorder have a GAA mutation in both copies of FXN.
Ataxia is inherited as an autosomal recessive condition. Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same condition, one from each parent. If a person receives one normal gene and one altered gene, he will be a carrier and will not show symptoms.
- The risk of two carrier parents passing on the defective gene to their offspring is 25% in each pregnancy.
- The risk of having a carrier child is 50%.
- The chance for a child to receive normal genes from both parents and be genetically normal for that particular condition is 25%.
The risks are the same for men and women.
Treatment of Friedreich's ataxia
Since the disease is hereditary, the entire treatment process comes down to delaying the progression of the disease. In most cases, this allows the patient to lead an active lifestyle for a long time and avoid complications.
To treat Friedreich's ataxia, metabolic medications are prescribed, which come in three types:
- stimulators of mitochondrial respiratory function;
- antioxidants - drugs that slow down oxidation;
- cofactors of enzyme reactions.
Drugs that nourish the heart muscle and improve its metabolism may also be prescribed.
In some cases, it is necessary to take butolotoxin, a drug that eliminates muscle spasms.
Exercise therapy is considered an important link in treatment. Particular attention is paid to muscle training and coordination of movements. A properly selected set of exercises helps to get rid of painful sensations when moving.
Sometimes attributed to dietary nutrition. The principle of the diet is to limit the consumption of carbohydrates, an excess of which provokes symptoms.
4.Treatment
There is currently no etiopathogenetic therapy or means of prevention; all measures taken (even radical ones, for example, orthopedic surgery) are only palliative in nature and aimed at mitigating the dominant symptoms. The prognosis is generally unfavorable: the steady progression of the neurodegenerative process results in death within 15-20 years from manifest manifestations. However, in some cases, when the patient is under constant therapeutic control and it is possible to slow down the development of severe cardiac, pulmonary, and endocrine insufficiency, patients live to old age.