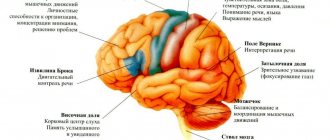
Причины атаксии Фридрейха
Данную патологию человек может получить только в случае, если оба его родителя будут носителями мутирующего гена. Мутация происходит в длинном плече девятой хромосомы, что и провоцирует нарушения в синтезе белка фратаксина из митохондрий, которые, в свою очередь, выполняют роль «клеточных энергетических станций».
В митохондриях накапливается железо и окисляется. Происходит транспорт кислорода в организме. При нарушении синтеза железа, его количество в митохондриях резко и значительно возрастает (примерно в десять раз). При этом клеточное железо остается в пределах нормы, а уровень цитозольного железа падает.
Такие процессы приводят в активацию гены, которые кодируют фрагменты, отвечающие за транспортировку железа — ферроксидаз и пермеаз. Баланс внутриклеточного железа нарушается еще больше. В результате высокой концентрации железа в клетке активизируются радикалы, которые обладают повреждающим действием и разрушают клетку изнутри. Самыми уязвимыми клетками оказываются нейроны (особенно в заднем и боковом столбах спинного мозга, в спиномозжечковых трактах, волокна периферических нервов).
Учитывая степень мутации гена, выделяются «классические» формы заболевания и атипичные, так сказать облегченные версии, доброкачественные синдромы.
Наследственная атаксия Фридрейха — самая распространенная среди атаксий.
1.Общие сведения
Атаксия (древнегреч. «неупорядоченность») – неврологический патологический феномен, заключающийся в рассогласованности, хаотичности моторики: нарушаются сложные нейромышечные взаимосвязи, в результате чего движения больного становятся нецеленаправленными и непродуктивными. Атаксия, выраженная в той или иной степени, сопутствует многим заболеваниям центральной нервной системы (ЦНС), – например, атрофическим и опухолевым, а также некоторым психопатологическим расстройствам, – однако выделяется группа атаксий как самостоятельных болезней.
В частности, известно более двух десятков вариантов наследственной спиноцеребеллярной (т.е. обусловленной сочетанным поражением спинного мозга и мозжечка) атаксии, или SCA, представляющей собой генетически обусловленный дегенеративно-дистрофический процесс в указанных структурах ЦНС.
Семейная атаксия Фридрейха (названа по имени врача, который в 1860 году дал первое клиническое описание болезни) является наиболее распространенным из наследственных нейродегенеративных заболеваний с преобладанием атаксии в клинической картине. Встречается, по различным источникам и в некоторой региональной зависимости, у 3-7 человек на каждые сто тысяч населения; пассивное носительство дефектного гена распространено гораздо шире (около 1%). От пола вероятность запуска процесса не зависит.
Обязательно для ознакомления! Помощь в лечении и госпитализации!
Симптомы атаксии Фридрейха
Клиническая картина более четко разворачивается в возрасте от 10 до 20 лет, хотя не исключено выявление симптоматики атаксии Фридрейха и в более позднем возрасте. Есть гипотеза, что классическая и атипичная формы данного заболевания могут быть вызваны различными мутациями одного или нескольких генов. Первые симптомы чаще всего проявляются в период становления репродуктивной системы.
Клиническая картина характеризуется сочетанием неврологических и экстраневральных симптомов. До появления ДНК диагностики клиническая картина заболевания описывалась только в классической форме. Позднее ученые пришли к выводу, что спектр заболевания намного глубже, а распространённость выше, поэтому стали выделять стертые и атипичные формы атаксии Фридрейха.
Среди неврологических симптомов при атаксии Фридрейха выделяют:
- Чувство неловкости и неуверенности при ходьбе (появляется одним из самых первых симптомов), которое усиливается если человек находится в темноте. Наблюдается пошатывание, человек часто спотыкается, случаются немотивированные падения. Наблюдается неустойчивость в позе Ромберга, невозможность повторить коленно-пяточную пробу. Со временем появляется дезориентация в руках, ноги быстро устают, может измениться почерк. При вытянутых руках наблюдается тремор, выполнение пальценосовой пробы невозможно (пациент постоянно промахивается).
- Дизартрия (может наблюдаться не во всех случаях), нарушения речевого аппарата.
- Происходит нарушение или полное исчезновение сухожильных и надкостничных рефлексов (наблюдается уже в раннем периоде клинической картины, является важным диагностическим звеном).
- Угнетение ахилловых и коленных рефлексов (иногда проявляются задолго до возникновения других признаком заболевания).
- Тотальная арефлексия (чаще происходит в развернутой стадии).
- Нарушение суставно-мышечной и вибрационной чувствительности.
- Симптом Бабинского (разгибание большого пальца как реакция на болевое раздражение стопы) — один из наиболее ранних признаков заболевания.
- Мышечная гипотония.
- Слабость ног и снижение мышечного тонуса может смениться полной атрофией.
- Мозжечковая и сенситивная атаксия.
- Со временем может наступить атаксия рук, амиотрофия, глубокое расстройство чувствительности, происходит распад моторных функций, что в конечном итоге приводит к невозможности самообслуживания.
- Нистагм (дрожание века), атрофия зрительных и слуховых нервов, умственная слабость при отсутствии адекватного лечения может наблюдаться нарушение функции тазовых органов.
К экстраневральным симптомам относятся:
- Поражение сердца.
- Нарастающая гипертрофическая или дилатационная кардиомиопатия (боли в области сердца, учащенное сердцебиение, частая одышка, даже при незначительных нагрузках, систолический шум в сердце). Часто именно кардиомиопатия как сопутствующее заболевание является причиной летального исхода при атаксии Фридрейха.
Нередко электрокартографическая симптоматика значительно опережает неврологические признаки атаксии Фридрейха (иногда на несколько лет), поэтому бывает очень сложно правильно диагностировать данное заболевание. Пациенты чаще всего длительное время стоят на учете у кардиолога с диагнозом ревмокардит.
Важными для диагностики признаками данного заболевание являются также скелетные деформации:
- выраженный сколиоз;
- стопа Фридрейха (свод стопы высоко вогнут, пальцы ног переразгибаются в основных фалангах и согнуты в дистальных);
- кифосколиоз;
- пальцы верхних и нижних конечностей деформированы.
Такие признаки, как и кардиомиопатия, могут появиться задолго до неврологических симптомов.
При атаксии Фридрейха наблюдается расстройство эндокринной системы, которое может проявляться в виде следующих заболеваний:
- дисфункция яичников;
- сахарный диабет;
- инфантилизм;
- гипогонадизм.
Очень часто у пациентов с атаксией Фридрейха обнаруживается катаракта, поэтому ее также считают частью клинической картины данного заболевания.
Атаксия Фридрейха характеризуется быстрым прогрессированием и наращиванием симптоматики. Длительность заболевания зачастую составляет не более двадцати лет.
Выраженная клиническая картина атипичной атаксии Фридрейха наблюдается позже, чем в классической форме — примерно на третьем-пятом десятке жизни человека.
Течение происходит в более облегченной форме, чем при классической атаксии и исход заболевания более благоприятен:
- Пациент длительное время сохраняет возможность к самообслуживанию.
- Отсутствует развитие сахарного диабета.
- Нет парезов, сохраняются рефлексы.
Такие клинические случаи описываются под названием «поздняя атаксия Фридрейха» или «атаксия Фридрейха с сохраненными рефлексами».
Атаксия Фридрейха (АФ)
— аутосомно-рецессивное заболевание, т.е. больные дети рождаются у пары родителей, которые оба клинически здоровы, но являются носителями патологического гена. Заболевание поражает нейроны центральной и периферической нервной системы: преимущественно поражаются пучки Голля, в меньшей степени пучки Бурдаха, Флексига, Говерса, а так же, пирамидные пути, задние корешки, спинномозговые ганглии и периферические нервы,клетки коры мозжечка, базальные ганглии, кора головного мозга, проводящие пути спинного мозга. В других системах заболеванию подвергаются не менее важные клетки органов, это клетки миокарда, β — клетки островков Лангерганца в поджелудочной железе, клетки сетчатки и костных тканей. Вызывает прогрессирующую дегенерацию центральной и периферической нервной системы. Большинство больных гомозиготны; содержание мРНК у них так мало, что иногда она вообще не определяется (в отличие от здоровых лиц и носителей гена атаксии Фридрейха). АФ возникает одинаково часто и у мужчин, и у женщин, не возникает у представителей негроидной расы и почти не встречается у коренных азиатов. Названа в честь немецкого врача Николауса Фридрейха, который первым описал её в 1860 году. Является наиболее часто встречаемой из наследственных атаксий: частота встречаемости атаксии Фридрейха составляет около 2-7 случаев на 100 тысяч человек.
| Симптомы атаксии Фридрейха чаще появляются на первом, втором десятилетии жизни, изредка на третьем и четвертом десятилетии. Появляется неуверенность, пошатывание, спотыкание при ходьбе, частые падения, нарушается почерк из-за тремора, появляется дизартрия, слабость в ногах, нарушается слух. Исчезают сухожильные и надкостничные рефлексы (в первую очередь ахиловые и коленные). Иногда ранним симптомом может быть ревмокардит. Больные не выполняют пяточно-коленную пробу, появляется покачивание в позе Ромберга, которое усиливается при закрывании глаз, расстройства сидения. Симптом Бабинского. Часто нистагм. Постепенно нарушается глубокая чувствительность, нарастает мышечная атрофия, на начальных этапах более выражена на нижних конечностях, с течением болезни захватывает и верхние. Формируется тотальная арефлексия. Атрофируется зрительный нерв, развивается катаракта, что ведет к слепоте, нарушается функция тазовых органов, развивается деменция. Развиваются эндокринные нарушения: сахарный диабет, гипогонадизм, инфантилизм, дисфункция яичников. Кардиомиопатии. Скелетные деформации: искривление позвоночника, кифосколиоз, «стопа Фридрейха» (высокий вогнутый свод стопы с переразгибанием пальцев в основных фалангах и сгибанием в дистальных), деформация пальцев рук и ног, косолапость. Течение болезни неуклонно прогрессирующее, при отсутствии адекватного лечения, длительность болезни обычно не превышает 20 лет. Непосредственной причиной смерти могут быть сердечная и легочная недостаточность, инфекционные осложнения. В редких случаях при отсутствии сахарного диабета и сердечных нарушений, больные доживают до 70-80 лет. Прогноз благоприятнее у женщин: более 20 лет с начала заболевания живут 100% женщин и только 63% мужчин. |
Диагностика:
Компьютерная томография головного мозга, которая остается основной диагностикой атаксий при этом заболевании малоэффективна, т.к. обнаруживает изменения только на поздних стадиях. Удается обнаружить только слабую степень атрофии мозжечка на ранней стадии и атрофию полушарий, расширение стволовых цистерн, боковых желудочков и субарахноидального пространства обоих полушарий на более поздних стадиях. Ранняя диагностика атаксии Фридрейха производится с помощью МР-томографии, которая дает возможность обнаружить атрофию спинного мозга и уменьшение поперечного размера спинного мозга, особенно усиливающееся в каудальном направлении на развернутой стадии, и умерено выраженную атрофию моста, мозжечка и продолговатого мозга. На начальной стадии обязательно проводится электрофизиологическое исследование, при таких исследованиях устанавливается тяжесть поражения чувствительности нервов конечностей. Характерный для данного заболевания электронейромиографический паттерн заключается в отсутствии или значительном снижении амплитуды потенциалов действия чувствительных нервов конечностей, при сравнительно небольшом снижении скорости проведения импульса по двигательным нервам. Для полной диагностики проводят нагрузочные тесты толерантности к глюкозе (для исключения сахарного диабета), рентгеновское исследование позвоночника. На ЭКГ — нарушение ритма, инверсия зубцаТ, изменения проводимости, при эхокарднографии особенно часто отмечаются нарушения проводимости, вплоть до полной блокады, и гипертрофия межжелудочковой перегородки. В ряде случаев клинические и электрокардиографические симптомы поражения сердца иногда на несколько лет опережают появление неврологических нарушений. Больные длительно наблюдаются у кардиолога или участкового терапевта, чаще всего с диагнозом “ревмокардит”. Для оценки митохондриальных нарушений с помощью цитохимического метода наиболее целесообразным представляется определение активности ряда ферментов-дегидрогеназ лимфоцитов: сукцинатдегидрогеназы (СДГ),
Дифференцирование диагноза:
Схожая симптоматика характерна для опухолей мозжечка, наследственных обменных заболеваний: Gm 1- и Gm 2-ганглиозидоз и галактосиалидоз (для дифференциации проводится исследование активности β-галактозидазы и гексозаминидазы А), нейросифилиса, фуникулярного миелоза, наследственной атаксии при дефиците витамина Е, синдрома Бассена-Корнцвейга, наследственных обменных заболеваниях, таких как болезнь Краббе (дифф. — исследование фермента галактозилцерамидазы) и поздний вариант болезни Ниманна-Пика (для дифференциации определяют содержание сфингомиелинов в цереброспинальной жидкости, исследуют стернальный пунктат на наличие «пенистых» клеток), болезнь Луи-Бар (или иначе атаксия-телеангиэктазия: клинически она отличается наличием на коже телеангиэктазий (чрезмерное локальное расширение мелких сосудов, преимущественно прекапилляров и капилляров), отсутствием скелетных аномалий, частыми и тяжело протекающими инфекциями дыхательных путей, отсутствием или крайне низким уровнем IgA, высоким уровнем а-фетопротеина. На МРТ выявляется гипоплазия мозжечка, чаще его червя.), при рассеянном склерозе (дифференциальный диагноз обычно не вызывает затруднений, т.к. для рассеянного склероза нехарактерны такие симптомы, как сухожильная арефлексия, мышечная гипотония, амиотрофии, экстраневральные проявления, а также в связи с отсутствием при болезни Фридрейха ремиссий и очаговых изменений плотности вещества мозга при КТ и МР-томографии). Для дифференцирования заболевания проводится ДНК тестирование и медико-генетическое консультирование, исследование липидного профиля крови, анализ мазка крови на наличие дефицита витамина Е и акантоцитов. ДНК тестирование должно назначаться не только больному, но и родственникам для определения наследственности заболевания, это необходимо в целях профилактики, и назначения превентивной терапии. При молекулярно-генетическом обследовании пациентов с клинически типичными проявлениями АФ на увеличение тринуклеотида ГАА расширение аллеля обнаруживается не у всех. При этом возможна точечная мутация или делеция в гене БФ на обеих хромосомах. В этих случаях это может быть фенокопия АФ, так как увеличение триплета было описано у многих больных атипичной атаксией и у пациентов с генерализованной хореей.
Лечение
атаксии Фридрейха не приводит к полному выздоровлению, но своевременная профилактика дает возможность избежать развития многих симптомов и осложнений. Для замедления прогрессирования заболевания назначаются препараты митохондриального ряда, антиоксиданты и другие препараты, уменьшающие аккумуляцию железа в митохондриях. Общий принцип лечения данными препаратами состоит в сочетанном назначении лекарств, синергично влияющих на разные уровни энергетического метаболизма. Рекомендуется одновременное назначение как минимум трех лекарственных средств из первых трех групп. (Препараты, повышающие активность дыхательной цепи митохондрий, кофакторы энзимных реакций энергетического обмена, антиоксиданты).Назначаются такие антиоксиданты, как витамины А и Е, а также синтетический заменитель коэнзима Q 10 – идебенон, который затормаживает нейродегенеративный процесс и развитие гипертрофической кардиомиопатии. Обычно назначают препараты, улучшающие метаболизм миокарда: рибоксин, кокарбоксилазу, предуктал и др. Назначается также 5-гидроксипрофан, который дает, неплохие результаты, но требует дальнейших исследований. В целом лечение симптоматическое, направлено на такие симптомы, как сахарный диабет, заболевания сердечно-сосудистой системы. Проводится общеукрепляющее лечение (витамины), а также препараты, влияющие на тканевый обмен (пирацетам, аминалон, ацефен, церебролизин), лечение которыми следует периодически повторять. Проводится также хирургическая коррекция стоп и введение ботулотоксина в спастичные мышцы. Физиотерапия и лечебная физкультура – процедуры, без которых лечение атаксии Фридрейха чаще всего оказывается малоэффективным. Постоянные занятия дают возможность держать тело в тонусе и устранить болезненные ощущения. Дети с АФ могут оставаться активными максимально долго, занимаясь лечебной физкультурой и комплексами корректирующих упражнений, которые следует фокусировать на тренировку баланса и силы мышц. При такой программе упражнений не развивается кардиомиопатия. Пациенты чувствуют себя лучше при ограничении углеводов в пище до 10г/кг, поскольку высокое их потребление является своеобразной «провокацией», усиливающей дефект энергетического обмена. Пациентам требуется социальная адаптация, так как многим приходится жить в состоянии полной беспомощности. Потеря зрения, возможности самостоятельно двигаться, нарушенная координация создает психологические нарушения, которые должны устраняться с помощью специалистов и поддержки близких.
Профилактика атаксии Фридрейха
— Особое значение имеет ДНК тестирование на ранней пресимптомной стадии с целью назначения превентивной терапии. Обследуются в первую очередь родственники больного.
Распространенность аутосомно-рецессивной атаксии Фридрейха среди якутского населения PC (Я) составляет 2,8 на 100 тыс. населения. Молекулярно-генетической причиной АФ у якутов является экспансия (GAA) n- повторов в 1 интроне гена FRDA. В улусах вилюйской и центральной Якутии зарегистрированы единичные случаи аутосомно-рецессивной атаксии Фридрейха без накопления в отдельных улусах. Интересен тот факт, что заболевание зарегистрировано у якутского этноса, являющегося представителем азиатской расы, среди которых ранее не было зарегистрировано случаев атаксии Фридрейха.. Это может быть объяснено привнесением в генофонд якутов европейской компоненты, поэтому выяснение причин возникновения и распространения атаксии Фридрейха среди якутов требует дальнейшего изучения.
Литература:
- ДНК-диагностика атаксии Фридрейха с клинико-генетическим анализом.// В. В. Пугачев, С. Н. Иллариошкин, Л. В. Прокутша, Б. Д. Маркова, 0. В. Евграфов, И. А. Ишнова-Смоленская // Сборник «Молекулярная диагностика наследственных заболеваний и медико-генетическое консультирование. «- М — MOlIHKll — 1QS3.
- Наследственные атаксии и параплегии // С.Н. Иллариошкин. // «Медицина», 2006.
- Наследственные болезни нервной системы.// Ю.Е. Вельтищев, П.А.Темин. // «Медицина», 1998.
- Неврология детского возраста // А.С. Петрухин. // «Медицина», 2004.
- Неврология. Справочник практического врача // Д.Р.Штульман, О.С. Левин // Издательство «МЕДпресс-информ», 2005г.
- Обнаружение полиморфного маркера в области гена, ответственного за возникновение атаксии Фридрейха. // О. В. Евграфов, В. В. Пугачев, Л Б. Стрельченко. // 2-ой Всесоюзный симпозиум «Теоретические и прикладные аспекты молекулярной биологии». Самарканд. — 1991.- тезисы дом. — С. 68.
- Поиск и изучение микросателлитных повторов в области предполагаемой локализации гена FRDA // О.В. Евграфов, В. В. Пугачев, А.В.Поляков, Л Б. Стрельченко // II съезд ВОГИС, Минск, 1992, Тезисы докл. ч.1.с.25
Компьютерная томография
Отсутствие адекватного лечения ускоряет процесс прогрессирования заболевания и приводит его в тяжелую стадию. Основным диагностическим методом всех атаксий считается компьютерная томография головного мозга. Но в данном случае она малоэффективна, так как большинство изменений в мозге при атаксии Фридрейха обнаруживаются только на поздних стадиях. Это объясняется спинальной локализацией изменений. Ранние стадии заболевания не видны при КТ. Зачатую в поздних стадиях можно обнаружить только незначительные атрофии мозжечка и полушарий, некоторое расширение мозговых цистерн, боковых желудочков, субарахноидального пространства.
Лечение заболевания
Адекватное и регулярной лечение атаксии Фридрейха позволяет приостановить прогрессирование заболевания, избежать осложнений, длительное время сохранять способность пациента вести активный образ жизни.
Как правило, атаксия Фридрейха лечится одновременным назначением метаболических препаратов, принадлежащих к 3 различным группам: кофакторов энергетических энзимных реакций, стимуляторов активности дыхательной цепи митохондрий и антиоксидантов.
Дополнительно при атаксии Фридрейха назначаются медикаменты, улучшающие метаболические процессы в сердечной мышце (кокарбоксилаза, рибоксин, предуктал, 5-гидроксипрофан и пр), ноотропы и нейропротекторы (аминалон, пирацетам, ацефен, церебролизин, энцефабол), поливитамины.
Большое значение для пациентов с атаксией Фридрейха имеет массаж и лечебная физкультура. Постоянные занятия ЛФК, направленные на тренировку координации и мышечной силы, дают возможность сохранять двигательную активность и купировать возникающие болезненные ощущения. Дети с АФ могут оставаться активными максимально долго, занимаясь лечебной физкультурой и комплексами корректирующих упражнений, которые следует фокусировать на тренировку баланса и силы мышц. При такой программе упражнений не развивается кардиомиопатия.
Хороший эффект оказывают такие физиотерапевтические процедуры, как тепловые процедуры (озокерит, парафин) на область предплечий и голеней, электростимуляция мышц.
Поскольку атаксия Фридрейха сопровождается нарушением энергетического обмена, то пациентам с этим заболеванием необходимо ограничить прием углеводов с пищей, избыток которых может провоцировать усугубление обменных нарушений.
Человек, страдающий от атаксии Фридрейха нуждается в некоторых хирургических вмешательств (в основном для позвоночника и сердца). Титановые винты и стержни вставляются в позвоночник, чтобы помочь предотвратить или замедлить прогрессирование сколиоза. Целью операции является сохранение состояния пациента как можно дольше. Во многих случаях у пациента развиваются болезни сердца. Эти болезни поддается медикаментозному лечению.
В некоторых случаях проводятся хирургическая коррекция деформации стоп, введение ботулотоксина в спастичные мышцы.
Больные нуждаются в социальной адаптации, так как многим приходится жить в состоянии полной беспомощности. Потеря зрения, возможности самостоятельно двигаться, нарушенная координация создает психологические нарушения, которые должны устраняться с помощью специалистов и поддержки близких.
Для остановки прогрессирования атаксия используют вспомогательные устройства, такие как трость, ходунки, или инвалидную коляску. Реабилитационные меры включают ряд физических и лечебных нагрузок.
МР-томография
Назначается МР-томография, с помощью которой удается обнаружить атрофию в спинном мозге на ранних стадиях, а также исследуются поперечные размеры спинного мозга. При атаксии Фридрейха они ниже нормы. Видна также умеренно выраженная атрофия моста, мозжечка и продолговатого мозга.
С помощью электрофизиологического исследования устанавливается степень поражения чувствительности нервов конечностей. При атаксии Фридрейха значительно снижена или вовсе отсутствует амплитуда потенциалов действия чувствительности нервов конечностей.
Симптомы
Первичным симптомом синдрома Фридрейха является прогрессирующая атаксия конечностей во время ходьбы. Атаксия предполагает неадекватную координацию мышц, что приводит к нестабильной походке, плохому контролю тонких движений конечностей.
Вовлечение мышц рта, горла может привести к невнятной речи, нарушению глотания. Интеллект, как правило, не затрагивается.
Могут развиться боковая кривизна позвоночника (сколиоз) или аномалии стопы.
Форма сердечной болезни (кардиомиопатия) присутствует у более половины людей с расстройством.
Поскольку нет эффективной терапии, клинические признаки прогрессируют, люди нуждаются в использовании инвалидной коляски для мобильности.
Дифференциальная диагностика
Для объективности диагноза пациент обязательно проходит консультацию нескольких врачей: эндокринолога, офтальмолога, ортопеда, кардиолога.
Диагностика этого генетического заболевания — непростой процесс из-за сложности дифференцировать заболевание в ряде других, практически идентичных, а часто и сопутствующих заболеваний:
- Наследственная атаксия по поводу нехватки витамина Е. Для дифференцирования определяют концентрацию витамина Е в крови, исследуют липидный профиль крови, выявляют наличие акантоцитоза с помощью мазка крови.
- Синдром Бассена—Корнцвейга.
- Заболевания, связанные с нарушением обмена веществ, которые наследуются по аутосомно-рецессивному типу. Например, болезнь Краббе, болезнь Ниманна-Пика.
- Рассеянный склероз.
Причины
Ген, ответственный за расстройство – FXN. Он кодирует фратаксин (frataxin), белок, необходимый для правильного функционирования митохондрий, которые производят энергию для клеток. Поскольку обе копии гена являются ненормальными, они не продуцируют достаточное количество белка frataxin.
Узнать больше Признаки диагностика и лечение синдрома Ротора
Ткани, которые особенно зависят от производства клеточной энергии – нервные, сердечные клетки начинают дегенерировать.
У большинства пораженных людей ген FXN содержит очень специфический тип ошибки, называемый расширенным повторением тринуклеотида GAA.
Каждый ген состоит из четырех химических единиц (нуклеотидов), называемых аденином (А), цитозином (С), гуанином (G), тимином (Т).
У большинства людей с расстройством обе копии гена FXN содержат аномально длинные участки повторяющихся звеньев, состоящие из гуанин-аденин-аденина (повтор тринуклеотида GAA). Здоровые люди имеют менее 30 повторов GAA, люди с FRDA получают расширенные тракты от 100 до 1300 повторов на обеих копиях гена FXN. Большинство содержит> 400 повторов.
Несмотря на то, что эта расширенная повторная мутация GAA находится в «некодирующей» области FXN (интрон 1) приводит к замораживанию генов и снижению способности продуцировать белок фратаксина. Тяжесть болезни и изменчивость симптомов пропорциональна длине расширенной повторной мутации.
Например, более короткие повторы (<400 GAA) связаны с более поздним возрастом начала, медленным прогрессированием клинических признаков, отсутствием или легкой кардиомиопатией.
Родители детей с синдромом Фридрейха имеющие одну копию повторной мутации GAA и один нормальный ген FXN не проявляют никаких признаков заболевания.
Большинство людей с расстройством имеют мутацию GAA в обеих копиях FXN.
Атаксия наследуется как аутосомно-рецессивное состояние. Рецессивные генетические нарушения возникают, когда индивидуум наследует две копии аномального гена одного состояния, по одному от каждого родителя. Если человек получает один нормальный ген и один измененный, то будет носителем и не проявит симптомов.
- Риск того, что два родителя-носителя передадут дефектный ген потомству, составляет 25% при каждой беременности.
- Риск иметь ребенка – носителя – 50%.
- Шанс для ребенка получить нормальные гены обоих родителей и быть генетически нормальным для этого конкретного состояния составляет 25%.
Риски одинаковы для мужчин и женщин.
Лечение атаксии Фридрейха
Так как заболевание наследственное, то весь процесс лечения сводится к задержке прогрессирования болезни. Это в большинстве случаев позволяет пациенту длительное время вести активный способ жизни и избегать осложнений.
Для лечения атаксии Фридрейха назначают прием метаболических лекарственных препаратов, которые бывают трех видов:
- стимуляторы дыхательной функции митохондрий;
- антиоксиданты — препараты, замедляющие окисление;
- кофакторы энзимных реакций.
Могут назначаться также препараты, которые питают сердечную мышцу и улучшают ее метаболизм.
В некоторых случаях необходим прием бутолотоксина — препарата, устраняющего мышечные спазмы.
Важным звеном в лечении считается ЛФК. Особое внимание уделяется тренировкам мышц и координации движений. Правильно подобранный комплекс упражнений помогает избавиться от болезненных ощущений при движении.
Иногда приписывается диетическое питание. Принцип диеты заключается в ограничении употребления углеводов, переизбыток которых провоцирует симптоматику.
4.Лечение
Этиопатогенетической терапии или средств профилактики на сегодняшний день нет; все принимаемые меры (даже радикальные, например, ортопедическое хирургическое вмешательство) носят лишь паллиативный характер и направлены на смягчение доминирующей симптоматики. Прогноз, в целом, неблагоприятный: неуклонное прогрессирование нейродегенеративного процесса результирует летальным исходом через 15-20 лет от манифестных проявлений. Однако в отдельных случаях, когда пациент находится под постоянным терапевтическим контролем и удается затормозить развитие тяжелой сердечной, легочной, эндокринной недостаточности, больные доживают до старости.