Полезная информация о других различных заболеваниях, которые начинаются на букву «Д»: Дебильность, Двухволновый вирусный менингоэнцефалит, Деменция с тельцами Леви, Депрессивный невроз, Дермальный синус, Детский церебральный паралич, Джексоновская эпилепсия, Диастематомиелия (дипломиелия), Дискогенная миелопатия, Дистрофическая миотония Россолимо-Штейнерта-Куршмана, Диабетическая энцефалопатия, Дисциркуляторная энцефалопатия, Диффузное аксональное повреждение головного мозга, Доброкачественная роландическая эпилепсия.
Понятие — дистрофическая миотония Россолимо-Штейнерта-Куршмана
Дистрофическая миотония Россолимо-Штейнерта-Куршмана — заболевание наследственного характера, обладающее медленной степенью развития, которое вызывает образование миотонии в комбинации с деформациями мышечных зон дистрофической природы. В основе представленного недуга находится повреждения миотонин-протеинкиназы. Описываемая болезнь образует в теле больного спазмы, атрофические проблемы мышечной ткани шейного отдела, лицевой области и дистальных районов конечностей, а также пониженный уровень интеллектуальный способностей, аритмию и эндокринологические изменения.
В качестве диагностических процедур специалист используют разнообразные клинические приемы, например: генеалогическое тестирование и анализ ДНК. Для лечения симптомов патологического процесса используются фармакологические средства, направленные против симптоматики отклонения и дальнейшего развития дистрофии мышечной ткани (применяются анаболические стероидные препараты, АТФ и прочее).
Краткие факты
Дистрофическая миотония Россолимо-Штейнерта-Куршмана — наследственный аномальный процесс, который способен попадать в организм ребенка от родителей через аутосомно-доминантный путь. В возрасте от 10 до 20 лет может распространяться классический тип болезни. В редких ситуациях может встречаться врожденная форма недуга, так как ее симптоматика заметна сразу после появления новорожденного на свет. Согласно морфологическим показаниям при описываемом заболевании выделяется комбинация гипертрофических деформаций конкретных мышечных районов вместе с атрофией других участков. Помимо этого, происходит замещение элементов мышечных частиц жировой и соединительной тканью. При исследовании с помощью электромикроскопа разных образцов мышечной ткани можно увидеть деструкцию миофибрилл и нарушения размеров митохондрий.
Открытие и суть заболевания
Россолимо, Штейнерт и Куршман изучали болезнь, являющуюся генетической патологией с аутосомно-доминантным типом наследования. Это значит, что один родитель имеет мутантный ген, больные дети при этом рождаются с вероятностью 50%. Заболевание носит характер семейного недуга и передается последующим поколениям по вертикали.
Сыновья и дочери в таких семьях болеют с одинаковой частотой, примерно 3 — 5 человек на 100 тысяч населения. Возраст начала заболевания, а также выраженность симптомов отличаются значительной вариабельностью.
Описаны ранние неонатальные и поздние формы, однако чаще всего заболевание дебютирует на втором, реже — на третьем десятке жизни. Отмечено, что передача болезни ребенку от матери является более прогностически неблагоприятной, чем от отца.
В основе болезни лежит дефект гена из 19 пары хромосом, который отвечает за синтез фермента миотонин-протеинкиназы. Это белок в норме присутствует не только в скелетной мускулатуре, но и в клетках миокарда и ЦНС.
Вот почему для дистрофической миотонии характерна полисистемность проявлений с поражением разных органов и систем. Неполноценность миотонин-протеинкиназы приводит к появлению мышечных спазмов вместе с атрофическими изменениями мускулатуры головы, шейного отдела, конечностей.
Наблюдается сочетание гипертрофии одних мышечных волокон с атрофией других и заменой их на жировую или соединительную ткани.
Основные источники появления миотонии Россолимо-Штейнерта-Куршмана
Согласно результатам последних проведённых тестирований генов пациентов с дистрофическим недугом указали на то, что основную роль в патологии играет дефективное расстройство, формирующееся в конкретном геноме, который располагается в девятнадцатой хромосоме. Именно она отвечает за синтезацию миотонин-протеинкиназы. У людей, страдающих описываемым отклонением от нормы, диагностируют серьезное возрастание в размерах некоторых частей гена под названием DMPK. Следует учитывать, что именно от количества повторов данного отдела зависит степень и разновидность описываемой болезни.
Ориентируясь на принятые нормы количество необходимых оборотов, колеблется в районе 5-37. В случае возрастания свыше 50-80, свидетельствует о наличии слабого проявления пагубного процесса, при увеличении показателей в пределе 100-500 — начинается поздняя стадия миотонии, а врожденный тип развивается при отклонении в 500-2000. Многие анализы показывают, что повышение уровня тринуклеотидных повторов прогрессирует, зачастую, в женских клетках на стадии мейоза. Именно поэтому во время передачи недуга от женщины проявляется максимальная тяжелая разновидность заболевания или ее врожденная форма.
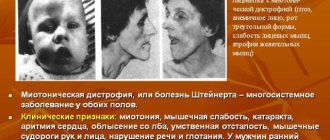
Миотоническая дистрофия (болезнь Штейнерта, дистрофическая миотония)
Миотоническая дистрофия — аутосомно-доминантное многосистемное заболевание, характеризующееся сильно вариабельной экспрессией гена (клиническим полиморфизмом) у обоих полов по началу заболевания и тяжести течения. Главные клинические проявления: миотония, мышечная слабость, катаракты, аритмии сердца, облысение со лба, нарушенная толерантность к глюкозе, умственная отсталость. Мышечные судороги особенно выражены на руках, челюстях, языке (в виде фибрилляции). Одновременно отмечается постепенно усиливающаяся мышечная слабость в связи с дегенерацией отёчных мышечных клеток и атрофией волокон. Миотония и мышечная слабость у пациентов сочетаются с нарушением речи и глотания. Начальные признаки миотонической дистрофии варьируют. Миотония вначале выявляется только при специальном тестировании. Мышечные подёргивания и слабость обычно асимметричны. В первую очередь в патологический процесс вовлекаются лицевые и височные мышцы (статус миотонического лица), затем — шейные, плечевые, бедренные мышцы от проксимального направления к дистальному.
Наряду с нервно-мышечными симптомами при миотонической дистрофии отмечаются катаракты (очень ранний симптом), гипогонадизм (атрофия семенников), аменорея, дисменорея, кисты яичника, облысение со лба (особенно у мужчин), изменения проводимости сердца с аритмией, абдоминальные симптомы (на почве холелитиаза), прогрессирующая умственная отсталость.
Тяжесть клинических проявлений очень сильно различается даже в пределах одной семьи.
Миотоническая дистрофия характеризуется варьирующим началом заболевания: от пренатального периода до 50-60 лет. Различают 4 формы по возрастному «пику» начала заболевания: врождённая, юношеская, классическая (20-30 лет) и минимальная (50-60 лет). Это объясняется различиями в числе тринуклеотидных повторов в локусе миотонической дистрофии.
Смерть при миотонической дистрофии наступает в возрасте 50-60 лет (при классической форме) как следствие пневмонии, сердечных осложнений или других интеркуррентных заболеваний. Частота болезни, по-видимому, не различается в этносах и популяциях, хотя эффект родоначальника описан у канадцев французского происхождения. Обобщённо распространённость миотонической дистрофии можно оценить как 1:7500-1:10 000.
Генетика миотонической дистрофии хорошо изучена на генеалогическом, формально-генетическом и молекулярно-генетическом уровнях. У пациентов всех стран обнаружена одна и та же мутация в гене протеинкиназы мышечной дистрофии (символ гена DM-PK
), локализованном в хромосоме 19ql3.3. Суть мутации — экспансия (увеличение числа) нестабильных CTG повторов в З’-нетранслируемой области гена. В норме число CTG повторов колеблется от 5 до 30. При миотонической дистрофии этот показатель значительно увеличивается и варьирует от 50 до 2500 и выше. Обнаружена корреляция между тяжестью течения и числом тринуклеотидных повторов. Чем больше повторов, тем раньше начинается заболевание и болезнь протекает тяжелее. Клиническая картина у гомозигот выражена в более тяжёлой форме.
Во многих семьях с миотонической дистрофией в нескольких поколениях отмечается антиципация, т.е. более тяжёлая манифестация болезни и в более молодом возрасте в каждом последующем поколении. Этот признак описан для миотонической дистрофии давно и рассматривался в 40-х годах как статистический артефакт. Однако сведения о молекулярном дефекте свидетельствуют о возможности увеличения числа триплетов в поколениях.
Описаны семьи более чем с тремя поколениями с миотонической дистрофией: в 1-м поколении — только катаракты, во 2-м поколении — умеренная слабость мышц, в 3-м поколении — врождённая форма.
При миотонической дистрофии выражен импринтинг. У пациентов, рождённых больными матерями, имеется более тяжёлая форма болезни с более ранним началом, чем у пациентов, рождённых от больных отцов. Врождённая форма миотонической дистрофии наблюдается только при рождении детей больными матерями. Механизм импринтинга выяснен: экспансия триплетов происходит в мейозе у женщин, а при сперматогенезе этот процесс отсутствует.
Уменьшение длины мутантного повтора (почти до нормы) у потомков с лёгкой клинической картиной и бессимптомным течением наблюдается при отцовской передаче гена. Одним из объяснений этого может быть селекция против длинных аллелей в мужском гаметогенезе.
В некоторых семьях с миотонической дистрофией обнаружена нормальная длина тринуклеотидного сегмента гена. Это можно объяснить двумя вариантами. Во-первых, точковая мутация в гене миотонинпротеинкиназы может нарушать транскрипцию/трансляцию или стабильность мРНК, т.е. прерывать синтез этого фермента, что и вызывает миотоническую дистрофию. Во-вторых, имеются доказательства локусной генетической гетерогенности миотонической дистрофии. Обнаружен второй локус «классического» дистального фенотипа миотонической дистрофии, картированный на хромосоме 3q.
В чем проявляется классическая дистрофическая миотония Россолимо-Штейнерта-Куршмана?
Стандартный аномальный процесс берет начало после пятилетнего возраста и может проявиться до 35-летнего периода, но чаще всего клиническая картина болезни появляется в промежутке между 10-ю и 20-ю годами. Симптоматика патологии является сочетанием проблем, возникающим при миотонии с факторами миопатии, нарушениями сердечно-сосудистой системы и ЦНС, а также нарушениями эндокринной структуры и катарактой.
Характерными признаками поражения организма считается формирование спазмов, локализующихся в жевательных мышцах и сгибательном районе кисти. Помимо этого, врачи диагностируют механические рефлексы миотонического плана, которые заметны при постукивании неврологическим инструментом. Главной особенностью миотонии Россолимо-Штейнерта-Куршмана принято называть присутствие атрофический нарушений в разных мышечных группах. В тоже время при дальнейшем прогрессировании отмечается постепенное снижение симптоматики вследствие развивающейся дистрофии мышц.
При расположении патологического процесса в зоне лицевой области у больного замечается маскообразная гримаса печального типа. Поражения мышечных тканей ротовой части дают путь к распространению парезов с деформацией голоса и трудностями при глотании. Изменения, характерны для миопатии, могут проявляться даже в дыхательной зоне, что вызывает снижение уровня легочной вентиляции, возникновению припадков апноэ во время сна, застойной или аспирационной пневмонии. Специалисты выделяют дополнительные жалобы пациентов:
- Проблемы в функционировании сердечно-сосудистой системы, фиксируется в 50% случаев.
- Расстройства ЦНС, способные привести больного к легкой форме дебильности.
- Эндокринные нарушения, влияют на работу половой системы — мужчины замечают низкую степень либидо, импотенцию, крипторхизм и т.д, а женщины — сбои менструации, ранний климакс и прочее.
- Деформация структуры волосяного покрова в комбинации с алопецией.
- У представителей мужского пола начинается облысение в височной и лобной зоне.
Международный неврологический журнал 3 (33) 2010
Миотония Куршманна — Баттена — Штейнерта, также известная под названиями «дистрофическая миотония I типа» (DM1), «атрофическая миотония Россолимо», составляет примерно от 92 до 98 % (по данным разных авторов) всех семей с дистрофической миотонией. В отечественной литературе впервые описана Россолимо в 1901 году; позднее, в 1909 году, H. Steinert и F.F. Batten дали подробное описание клинической картины заболевания. Лишь в 1992 г. был идентифицирован генетический дефект при DM1. Многие авторы считают эту патологию переходной между миотониями и миопатиями. Заболевание имеет аутосомно-доминантный тип наследования. Встречается в двух клинических формах: врожденной и классической.
Эпидемиология
Встречаемость дистрофической миопатии I типа — примерно 1 на 8000 новорожденных, а распространенность в мире варьирует от 2,1 до 14,3 на 100 000 населения. В настоящее время считается, что нет достоверных половых различий в заболеваемости. Выраженность симптомов, возраст, при котором дебютирует заболевание, отличаются значительной гетерогенностью. Описаны неонатальные и поздние формы, хотя наиболее часто заболевание дебютирует в возрасте 14–30 лет.
Этиопатогенез
Экспрессируемый патологическим геном белок миотонин-протеинкиназа является представителем семейства серин/треонин-протеинкиназ и состоит из 624 аминокислот. Традиционно считается, что этот белок играет существенную роль в регуляции клеточной дифференцировки и репликации ДНК. Выделяют три изоформы миотонин-протеинкиназы в скелетных мышцах, образующиеся в процессе сплайсинга. В настоящее время в качестве одного из наиболее вероятных патогенетических механизмов миотонической дистрофии рассматривается влияние СТG-повторов на посттранскрипционную модификацию мРНК гена. Наиболее вероятно, при увеличении числа CTG-повторов происходит изменение процессинга мутантного РНК продукта или транспорта мРНК из ядра в цитоплазму на различных этапах. Нарушение укладки нуклеосом из-за изменения локальной структуры хромосом, с наличием экспансии тринуклеотидных повторов, также является одним из патогенетических механизмов (табл. 1).
Особенности клинического течения
Для миотонии Куршманна — Баттена — Штейнерта — Россолимо характерно сочетание миотонического синдрома, сердечно-сосудистых нарушений, эндокринной патологии в виде полигландулярной недостаточности. Часто наблюдается развитие атрофий, вследствие чего становится характерным вид пациента: голова опущена на грудь, лицо гипомимичное, полуптоз, дистальные амиотрофии конечностей. Типичны «выеденные» стопы, «обезьяньи» кисти. Походка степпажная, иногда при атрофиях проксимальных групп мышц напоминает утиную. Мышечный тонус снижен, глубокие рефлексы рано угасают. Многообразны нейроэндокринные расстройства: наиболее выражены изменения в гонадах. У многих больных отмечаются раннее облысение, истончение и сухость кожи. Сердечно-сосудистые расстройства постоянны. Наиболее часто на ЭКГ отмечается полная или частичная блокада ножек пучка Гиса, низкий вольтаж, различные аритмии.
Ведущим в клинике миотонии Куршманна — Баттена — Штейнерта — Россолимо является миотонический синдром, который имеет следующие характерные особенности:
1) нарушение расслабления мышц после их форсированного сокращения (при обычных целенаправленных движениях);
2) повторные мышечные сокращения обычно сопровождаются уменьшением степени выраженности миотонического феномена;
3) миотонический феномен может проявляться возникновением характерного мышечного валика в месте перкуссии мышцы с ямкой в точке нанесения удара. Он объясняется нарастанием нестабильности мембран мышечных волокон, их деполяризацией;
4) проявления миотонии нарастают на холоде и уменьшаются в тепле, а также при повторных действиях.
Также для верифицирования миотонического синдрома используют следующие тесты:
1. Тест на миотонию — больному предлагают совершать повторные однотипные действия: например, в быстром темпе интенсивное сжатие пальцев в кулак и их распрямление. При миотонии после первого сжатия кистей возникает тонический спазм сгибателей пальцев, после чего распрямление кистей происходит замедленно, с трудом. При повторении тех же действий выраженность мышечного спазма постепенно уменьшается.
2. Миотоническая реакция — реакция мышцы на механическое или электрическое раздражение, характеризующаяся длительным сильным сокращением и последующим медленным расслаблением.
3. Миотонические рефлексы — происходит замедленное расслабление мышц, сокращающихся при вызывании сухожильных или кожных рефлексов.
4. Симптом мышечного валика — при ударе молоточком по мышце, в частности по языку, у больного с миотонией на месте удара некоторое время сохраняется ямка или валик, иногда ямка, окруженная валиком, которую можно наблюдать в течение нескольких секунд, в тяжелых случаях — до минуты.
5. Ложный симптом Грефе — больному предлагается посмотреть вверх, а затем быстро опустить взор. При миотонии глазные яблоки поворачиваются вниз. А верхние веки при этом отстают, и между верхним веком и краем радужки остается полоска склеры, как это бывает при симптоме Грефе. В случае миотонии повторение тех же действий ведет к уменьшению отставания верхнего века.
6. Симптом возвышения большого пальца — при ударе молоточком по возвышению большого пальца кисти происходит приведение этого пальца, продолжающееся от нескольких секунд до минут и обусловленное спазмом приводящей мышцы большого пальца.
7. Феномен миотонического спазма разгибателей кисти — удар молоточком по мышцам — разгибателям кисти на предплечье на 4–6 см ниже локтевого сустава вызывает быстрое разгибание кисти с последующим ее «застыванием» на несколько секунд, а затем медленным возвращением в исходное положение.
8. Феномен приседания — больной с миотонией при приседании обязательно становится на носки. Если он при этом пытается опираться на всю подошвенную поверхность стоп, сближая в то же время медиальные поверхности бедер и голеней, то становится неустойчивым и может упасть.
9. Миотонический генерализованный спазм — возникает спазм, охватывающий всю мускулатуру при внезапном резком движении или при попытке сохранить нарушенное равновесие. Больной при этом нередко падает и оказывается некоторое время обездвиженным. Спазм появляется при тяжелых формах миотонии.
Необходимо отметить, что диагностика синдрома дисфагии, встречающегося при миотонии Куршманна — Баттена — Штейнерта, представляет собой сложную задачу. Дифференциальную диагностику следует проводить между орофарингеальной и эзофагеальной формами дисфагии (табл. 2).
Лабораторная диагностика
Общеклинические анализы крови и мочи при данной патологии не являются диагностическим маркером. Сывороточная КФК обычно в норме или наблюдается повышение в несколько раз. Возможно снижение концентрации тестостерона и IgG в сыворотке крови. На ЭМГ (при использовании игольчатых электродов) выявляют миотонические или псевдомиотонические высокоамплитудные разряды, нарастающие по амплитуде при мышечном сокращении с последующим их уменьшением. Цикл между идентичными фазами биоэлектрической реакции мышцы занимает приблизительно 500 мс. Разряды сопровождаются характерным звуковым феноменом («звук пикирующего бомбардировщика»).
С учетом многообразия клинических синдромов диагностика миопатии Куршманна — Баттена — Штейнерта представляет собой сложную задачу. Приводим наблюдение.
Больной Е., 21 года, при поступлении жалуется на судорожные стягивания в руках и ногах, головную боль, тошноту, повышенную утомляемость.
Родился недошенным в сроке до 32 недель, находился на диспансерном учете у детского невролога, был признан инвалидом детства, в последующем — инвалидом III группы. В детстве выполнялась МРТ головного мозга — выявлена гипотрофия коры гемисферы лобно-теменных долей. Ранее обследовался по поводу перинатальной энцефалопатии с синкопальными состояниями, с ликворной дисциркуляцией. Наблюдался по поводу хронического аутоиммунного тиреоидита. Около 1 года назад стал отмечать боли в ногах. Неоднократно выполнялось МРТ LS-отдела позвоночника — выявлены артрозные изменения в дугоотростчатых сочленениях, остеохондроз дисков. Неоднократно проводилось электронейромиографическое (ЭНМГ) исследование (в детстве), с помощью которого выявлялись элементы, характерные для митохондриальной миопатии. Состояние пациента ухудшилось в последние 2 недели, на фоне физической нагрузки наросла интенсивность болей в мышцах ног, усилились судорожные стягивания в них, особенно при движении.
При объективном исследовании состояние относительно удовлетворительное. Кожные покровы и видимые слизистые оболочки бледные. В легких дыхание везикулярное, хрипов нет. Деятельность сердца ритмичная, тоны приглушены. АД — 110/60 мм рт.ст. Выраженное диспластическое телосложение, гипотрофия кистей, дисплазия мелких суставов пальцев рук.
В неврологическом статусе отмечаются экзофтальм справа, синдром Горнера слева, нистагм, слабость конвергенции с двух сторон. Легкий симптом Вюрпе. Симптом Маринеску — Радовичи, четче справа. Сухожильные рефлексы с конечностей высокие, равны. Отмечается замедленное расслабление мышц после вызывания сухожильных рефлексов. Впечатление псевдогипертрофии мышц бедер. Положительный симптом «мышечного валика» в m.biceps femoralis с двух сторон, симптом разгибателя большого пальца, ложный симптом Грефе. Положительный тест на миотонию. При подъеме вытянутых ног отмечаются крампи в нижней трети бедер. Вибрационная чувствительность 9–11 секунд. Симптом Штрюмпеля с двух сторон. Мышечная слабость в проксимальных отделах конечностей. S-образный сколиоз. Ограничение функционального объема движений в L-отделе позвоночника. Походка по типу «утиной».
При лабораторных клинических и биохимических исследованиях крови и мочи патологии не выявлено. КФК крови в норме. ЭНМГ — при игольчатом исследовании mm.deltoideus, mm.biceps brachii, mm.peroneus longus dextra et sinistra, extensor digiti communis sinistra, deltoideus dextra выявляется спонтанная активность, состоящая из потенциалов фибрилляций и положительных острых волн, в виде миотонических, убывающих разрядов. Миотонические разряды сопровождаются характерным звуком пикирующего самолета (рис. 1).
На МРТ головного мозга выявляется очаговое изменение в белом веществе лобной области справа, наиболее вероятно сосудистого генеза. Расширение желудочков го ловного мозга, цистерн, подоболочечных пространств. МР-признаки снижения кровотока по левой СМА в сегменте М2, по правой ЗМА в сегменте Р3. ЭКГ — ритм синусовый, брадикардия — 56 ударов в минуту, признаки диффузного изменения миокарда. Эхокардиографически выявлен пролапс митрального клапана 0–1-й степени с минимальной регургитацией.
Пациент получал кардонат, сирдалут, актовегин, цераксон, аевит, АТФ-лонг, глицин.
На фоне проводимого лечения состояние улучшилось, уменьшилась частота крампи. Сохраняется легкий тетрапарез, амиотрофический синдром в руках.
Таким образом, сочетание активных и механических миоклонических реакций, миопатического синдрома, признаков поражения сердечно-сосудистой системы, прогредиентное течение, а также признаки миотонии на ЭНМГ позволили нам поставить диагноз дистрофической миотонии Куршманна — Баттена — Штейнерта с легким тетрапарезом, частыми крампи, синкопальными состояниями в анамнезе, легкими нарушениями функции ходьбы, медленно прогрессирующее течение. Соп.: последствия интранатального поражения нервной и мышечной системы в виде энцефаломиелопатии, с ликворно-сосудистой дисциркуляцией, атрофическими изменениями, полигландулярной недостаточностью, митохондриальными нарушениями.
Для постановки диагноза миотонической дистрофии необходимо учитывать следующие критерии: аутосомно-доминантный тип наследования, ранний дебют заболевания, сочетание миотонического и миопатического симптомокомплексов, полисистемность поражения, в частности сердечно-сосудистой системы (кардиомиопатии, нарушения ритма и проводимости сердца), прогрессирующее течение. Обязательным является ЭНМГ-исследование. Терапевтический эффект оказывает комплексная терапия (в сочетании с сирдалутом, глицином).
Симптоматика врожденной формы миотонии Россолимо-Штейнерта-Куршмана
В тех ситуациях, когда неврологи диагностируют хронический вариант заболевания, первыми признаками наличия аномалии являются патологические процесс, возникшие еще во внутриутробном периоде. Зачастую, симптоматика проявляется в серьезном понижении двигательной деятельности развивающегося плода, которая замечается во время ультразвукового исследования доктором в сфере гинекологии и акушерства (в периоде третьего триместра беременности). После рождения у ребенка выделяют следующие проблемы:
- Диффузные нарушения мышечной ткани — локализуется в точках мимической, жевательной и глазодвигательных мышечных тканях, а также в мускулатуре дистальных участков конечностей.
- Проблемы при вскармливании.
- Сбои в функционировании дыхательного аппарата.
- Заторможенность прогрессирования моторики.
- Появление олигофрении.
Фиксируется повышенная детская смертность при увеличении симптомов недуга.
Клинические проявления
В связи с варьированием начала заболевания в клинике различают следующие формы по возрастному принципу:
- врожденная форма — манифестация болезни начинается сразу после появления ребенка на свет;
- юношеский вариант — дебют миотонии в возрасте от одного года до периода полового созревания;
- классическая форма — начало клинических проявлений приходится на второй и третий десяток жизни;
- минимальный вариант — манифестация приходится на поздние сроки — шестой десяток жизни.
Характерно, что чем позднее проявляется болезнь, тем благоприятнее течение и лучше прогноз. Чаще всего встречается классическая форма болезни Штейнерта, для которой типичными являются следующие клинические симптомы:
- Миотония — проявляется спазмами жевательных мышц и сгибателей кистей рук, характерны атрофические изменения в разных группах мышц. Постепенно происходит угасание миотонических проявлений и прогрессирования мышечной дистрофии, внешне это выражается в печальной маске лица и отсутствии мимики. Опасным является парез мышц гортани с нарушением глотания, а также слабость дыхательной мускулатуры, в результате возможны приступы остановки дыхания во сне, развитие пневмонии.
- Сердечнососудистые нарушения — нарушения ритма сердца, гипертрофические изменения левого желудочка, выявляемые на ЭКГ, застойная сердечная недостаточность.
- Эндокринные расстройства (в основном затрагиваются половые функции) — уменьшение размеров половых органов, снижение сексуального влечения, у женщин — расстройства менструального цикла, ожирение.
- Общие изменения дистрофического характера — сухость и пигментация кожных покровов, выпадение частично или полностью волос и зубов, ранняя катаракта.
- Нарушения со стороны ЦНС — усталость, расстройства сна, апатия, потеря интеллекта.
Отдельно стоит отметить характерные клинические проявления врожденной формы дистрофической миопатии:
- уменьшение активных движений плода в утробе матери, выявляемое во время УЗИ;
- в период новорожденности — вялость, распространенная гипотония, особенно в жевательных, мимических, мышцах глазных яблок;
- сохранение и даже повышение сухожильных рефлексов;
- проблемы вскармливания, расстройства дыхания по типу респираторного дистресс-синдрома;
- задержка физического и нервно-психического развития, признаки олигофрении;
- стремительное прогрессирование заболевания, высокий риск внезапной смерти.
Варианты диагностики
При наличии вышеперечисленных жалоб, необходимо в срочном порядке посетить квалифицированного специалиста в сфере неврологии. Для подтверждения заключения, врач предпишет прохождение определенных клинических процедур, например:
- Генеалогическое обследование, которое покажет присутствие аутосомно-доминантной формы наследования патологии.
- ДНК-тест.
- Электромиографическая диагностика.
- Электронейрографическое обследование.
- Проверка половых гормонов.
- Электрокардиографическая оценка.
Помимо этого проводятся консультации у докторов, специализирующихся на проблемах наследственного, кардиологического, эндокринологического, гинекологического и прочих типов.
Способы лечения дистрофической миотонии Россолимо-Штейнерта-Куршмана
В настоящее время ученые еще не смогли составить радиальный план для избавления от признаков описываемого заболевания. Больным, страдающим от миотонии дистрофического характера, назначается особое диетическое питание, которое включает низкое содержание калия в продуктах. Помимо этого, врачи предписывают такие процедуры:
- Не находится долгое время на холоде.
- Употребление хинина, фенитоина и прочих медикаментов.
- Прием стероидных фармакологических препаратов, обладающих анаболическими свойствами.
- Назначаются малые дозировки АТФ и витаминные комплексы, в частности витамины группы В.