Useful information about other various diseases that begin with the letter “D”: Debility, Double-wave viral meningoencephalitis, Dementia with Lewy bodies, Depressive neurosis, Dermal sinus, Cerebral palsy, Jacksonian epilepsy, Diastematomyelia (diplomyelia), Discogenic myelopathy, Dystrophic myotonia Rossolimo -Steinert-Kurshman, Diabetic encephalopathy, Dyscirculatory encephalopathy, Diffuse axonal brain damage, Benign rolandic epilepsy.
Concept - dystrophic myotonia Rossolimo-Steinert-Kurshman
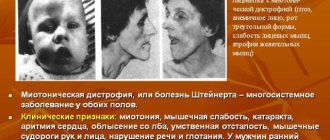
Dystrophic myotonia Rossolimo-Steinert-Kurshman is a hereditary disease with a slow development, which causes the formation of myotonia in combination with deformations of the muscle zones of a dystrophic nature. The basis of the presented disease is damage to myotonin-protein kinase. The described disease causes spasms in the patient’s body, atrophic problems in the muscle tissue of the cervical region, facial area and distal areas of the extremities, as well as a reduced level of intellectual abilities, arrhythmia and endocrinological changes.
As diagnostic procedures, specialists use a variety of clinical techniques, for example: genealogical testing and DNA analysis. To treat the symptoms of the pathological process, pharmacological agents are used to combat the symptoms of deviation and further development of muscle tissue dystrophy (anabolic steroid drugs, ATP, etc. are used).
Quick Facts
Dystrophic myotonia Rossolimo-Steinert-Kurshman is a hereditary abnormal process that can enter the child’s body from parents through an autosomal dominant pathway. Between the ages of 10 and 20, the classic type of the disease can spread. In rare situations, a congenital form of the disease may occur, since its symptoms are noticeable immediately after the birth of a newborn. According to morphological indications, the disease described is characterized by a combination of hypertrophic deformations of specific muscle areas along with atrophy of other areas. In addition, elements of muscle particles are replaced by fatty and connective tissue. When examining different samples of muscle tissue using an electromicroscope, one can see the destruction of myofibrils and disturbances in the size of mitochondria.
Discovery and essence of the disease
Rossolimo, Steinert and Kurshman studied the disease, which is a genetic pathology with an autosomal dominant mode of inheritance.
This means that one parent has a mutant gene, and sick children are born with a 50% probability. The disease is a family illness and is transmitted vertically to subsequent generations. Sons and daughters in such families get sick with the same frequency, approximately 3 - 5 people per 100 thousand population. The age at onset of the disease, as well as the severity of symptoms, vary widely.
Early neonatal and late forms have been described, but most often the disease debuts in the second, less often in the third decade of life. It has been noted that transmission of the disease to a child from the mother is more prognostically unfavorable than from the father.
The disease is based on a gene defect from the 19th pair of chromosomes, which is responsible for the synthesis of the enzyme myotonin-protein kinase. This protein is normally present not only in skeletal muscles, but also in myocardial and central nervous system cells.
That is why dystrophic myotonia is characterized by polysystemic manifestations affecting different organs and systems. Inferiority of myotonin-protein kinase leads to the appearance of muscle spasms along with atrophic changes in the muscles of the head, cervical region, and limbs.
There is a combination of hypertrophy of some muscle fibers with atrophy of others and their replacement with adipose or connective tissue.
The main sources of Rossolimo-Steinert-Kurshman myotonia
According to the results of recent testing of the genes of patients with dystrophic disease, it was indicated that the main role in the pathology is played by a defective disorder formed in a specific genome, which is located on the nineteenth chromosome. It is responsible for the synthesis of myotonin-protein kinase. People suffering from this abnormality are diagnosed with a severe increase in the size of certain parts of a gene called DMPK. It should be borne in mind that the degree and type of the disease being described depends on the number of repetitions of this section.
Based on accepted standards, the number of required revolutions fluctuates around 5-37. In the case of an increase above 50-80, it indicates the presence of a weak manifestation of a harmful process; with an increase in indicators in the range of 100-500, the late stage of myotonia begins, and the congenital type develops with a deviation of 500-2000. Many analyzes show that the increase in the level of trinucleotide repeats progresses, often in female cells at the stage of meiosis. That is why during the transmission of the disease from a woman, the maximum severe type of the disease or its congenital form appears.
Myotonic dystrophy (Steinert's disease, dystrophic myotonia)
Myotonic dystrophy is an autosomal dominant multisystem disease characterized by highly variable gene expression (clinical polymorphism) in both sexes in terms of disease onset and severity. Main clinical manifestations: myotonia, muscle weakness, cataracts, cardiac arrhythmias, baldness from the forehead, impaired glucose tolerance, mental retardation. Muscle cramps are especially pronounced in the arms, jaws, and tongue (in the form of fibrillation). At the same time, gradually increasing muscle weakness is noted due to degeneration of edematous muscle cells and fiber atrophy. Myotonia and muscle weakness in patients are combined with speech and swallowing disorders. The initial signs of myotonic dystrophy vary. Myotonia is initially detected only with special testing. Muscle twitching and weakness are usually asymmetrical. First of all, the facial and temporal muscles are involved in the pathological process (myotonic face status), then the cervical, shoulder, and femoral muscles from the proximal to the distal direction.
Along with neuromuscular symptoms in myotonic dystrophy, cataracts (a very early symptom), hypogonadism (testicular atrophy), amenorrhea, dysmenorrhea, ovarian cysts, baldness from the forehead (especially in men), changes in cardiac conductivity with arrhythmia, abdominal symptoms (due to cholelithiasis), progressive mental retardation.
The severity of clinical manifestations varies greatly, even within the same family.
Myotonic dystrophy is characterized by a variable onset of the disease: from the prenatal period to 50-60 years. There are 4 forms according to the age “peak” of the onset of the disease: congenital, juvenile, classic (20-30 years) and minimal (50-60 years). This is explained by differences in the number of trinucleotide repeats in the myotonic dystrophy locus.
Death with myotonic dystrophy occurs at the age of 50-60 years (with the classic form) as a result of pneumonia, cardiac complications or other intercurrent diseases. The incidence of the disease does not appear to vary across ethnic groups or populations, although a founder effect has been described in French Canadians. In general, the prevalence of myotonic dystrophy can be estimated as 1:7500-1:10,000.
The genetics of myotonic dystrophy has been well studied at the genealogical, formal genetic and molecular genetic levels. Patients from all countries have the same mutation in the muscular dystrophy protein kinase gene (gene symbol DM-PK
), localized on chromosome 19ql3.3. The essence of the mutation is the expansion (increase in the number) of unstable CTG repeats in the 3'-untranslated region of the gene. Normally, the number of CTG repeats ranges from 5 to 30. With myotonic dystrophy, this figure increases significantly and varies from 50 to 2500 and above. A correlation was found between the severity of the disease and the number of trinucleotide repeats. The more repetitions, the earlier the disease begins and the more severe the disease. The clinical picture in homozygotes is more severe.
In many families with myotonic dystrophy, anticipation is observed in several generations, i.e. more severe manifestation of the disease and at a younger age in each subsequent generation. This feature has been described for myotonic dystrophy for a long time and was considered in the 40s as a statistical artifact. However, information about the molecular defect indicates the possibility of increasing the number of triplets in generations.
Families with more than three generations with myotonic dystrophy have been described: in the 1st generation - only cataracts, in the 2nd generation - moderate muscle weakness, in the 3rd generation - a congenital form.
In myotonic dystrophy, imprinting is pronounced. Patients born to affected mothers have a more severe form of the disease with an earlier onset than patients born to affected fathers. The congenital form of myotonic dystrophy is observed only when children are born to sick mothers. The mechanism of imprinting has been clarified: expansion of triplets occurs in meiosis in women, but this process is absent during spermatogenesis.
A decrease in the length of the mutant repeat (almost to normal) in offspring with a mild clinical picture and an asymptomatic course is observed during paternal transmission of the gene. One explanation for this could be selection against long alleles in male gametogenesis.
Some families with myotonic dystrophy have a normal length of the trinucleotide gene segment. This can be explained in two ways. First, a point mutation in the myotonin protein kinase gene can impair transcription/translation or mRNA stability, i.e. interrupt the synthesis of this enzyme, which causes myotonic dystrophy. Second, there is evidence for locus genetic heterogeneity in myotonic dystrophy. A second locus for the “classic” distal myotonic dystrophy phenotype has been discovered, mapped to chromosome 3q.
What is the manifestation of classic dystrophic Rossolimo-Steinert-Kurshman myotonia?
The standard abnormal process begins after the age of five and can appear before the age of 35, but most often the clinical picture of the disease appears between the ages of 10 and 20. The symptoms of the pathology are a combination of problems that arise from myotonia with myopathy factors, disorders of the cardiovascular system and central nervous system, as well as disorders of the endocrine structure and cataracts.
Characteristic signs of damage to the body are the formation of spasms localized in the masticatory muscles and the flexor region of the hand. In addition, doctors diagnose mechanical reflexes of the myotonic type, which are noticeable when tapping with a neurological instrument. The main feature of Rossolimo-Steinert-Kurshman myotonia is usually called the presence of atrophic disorders in different muscle groups. At the same time, with further progression, there is a gradual decrease in symptoms due to developing muscle dystrophy.
When the pathological process is located in the facial area, the patient notices a mask-like grimace of a sad type. Lesions in the muscle tissue of the mouth give rise to the spread of paresis with voice deformation and difficulty swallowing. Changes characteristic of myopathy can even appear in the respiratory zone, which causes a decrease in the level of pulmonary ventilation, the occurrence of attacks of apnea during sleep, congestive or aspiration pneumonia. Experts highlight additional patient complaints:
- Problems in the functioning of the cardiovascular system are recorded in 50% of cases.
- Central nervous system disorders that can lead the patient to a mild form of debility.
- Endocrine disorders affect the functioning of the reproductive system - men notice low libido, impotence, cryptorchidism, etc., and women notice menstruation problems, early menopause, etc.
- Deformation of the hair structure in combination with alopecia.
- In males, baldness begins in the temporal and frontal areas.
International Neurological Journal 3 (33) 2010
Kurschmann-Batten-Steinert myotonia, also known as dystrophic myotonia type I (DM1), Rossolimo atrophic myotonia, accounts for approximately 92 to 98% (according to various authors) of all families with dystrophic myotonia. Rossolimo was first described in Russian literature in 1901; later, in 1909, H. Steinert and FF Batten gave a detailed description of the clinical picture of the disease. It was not until 1992 that the genetic defect in DM1 was identified. Many authors consider this pathology to be transitional between myotonia and myopathies. The disease has an autosomal dominant pattern of inheritance. It occurs in two clinical forms: congenital and classical.
Epidemiology
The incidence of dystrophic myopathy type I is approximately 1 in 8,000 newborns, and the worldwide prevalence varies from 2.1 to 14.3 per 100,000 population. It is currently believed that there are no significant sex differences in incidence. The severity of symptoms and the age at which the disease debuts are characterized by significant heterogeneity. Neonatal and late forms have been described, although the disease most often debuts at the age of 14–30 years.
Etiopathogenesis
The myotonin protein kinase protein expressed by the pathological gene is a member of the serine/threonine protein kinase family and consists of 624 amino acids. It is traditionally believed that this protein plays an essential role in the regulation of cell differentiation and DNA replication. There are three isoforms of myotonin protein kinase in skeletal muscles, formed during the splicing process. Currently, the influence of CTG repeats on post-transcriptional modification of gene mRNA is considered as one of the most likely pathogenetic mechanisms of myotonic dystrophy. Most likely, with an increase in the number of CTG repeats, a change occurs in the processing of the mutant RNA product or in the transport of mRNA from the nucleus to the cytoplasm at various stages. Disruption of nucleosome folding due to changes in the local structure of chromosomes, with the presence of expansion of trinucleotide repeats, is also one of the pathogenetic mechanisms (Table 1).
Features of the clinical course
Kurschmann-Batten-Steinert-Rossolimo myotonia is characterized by a combination of myotonic syndrome, cardiovascular disorders, and endocrine pathology in the form of polyglandular insufficiency. The development of atrophies is often observed, as a result of which the patient’s appearance becomes characteristic: the head is lowered to the chest, the face is hypomimic, hemiptosis, distal amyotrophies of the limbs. Typical are “eaten out” feet and “monkey” hands. The gait is stepping, sometimes with atrophy of the proximal muscle groups, it resembles a duck’s. Muscle tone is reduced, deep reflexes fade early. Neuroendocrine disorders are diverse: the most pronounced changes are in the gonads. Many patients experience early baldness, thinning and dry skin. Cardiovascular disorders are constant. Most often, the ECG shows complete or partial blockade of the bundle branches, low voltage, and various arrhythmias.
The leader in the clinic of Kurschmann-Batten-Steinert-Rossolimo myotonia is myotonic syndrome, which has the following characteristic features:
1) impaired muscle relaxation after forced contraction (during normal purposeful movements);
2) repeated muscle contractions are usually accompanied by a decrease in the severity of the myotonic phenomenon;
3) the myotonic phenomenon can be manifested by the appearance of a characteristic muscle ridge at the site of percussion of the muscle with a fossa at the point of impact. It is explained by an increase in instability of muscle fiber membranes and their depolarization;
4) manifestations of myotonia increase in the cold and decrease in warmth, as well as with repeated actions.
The following tests are also used to verify myotonic syndrome:
1. Test for myotonia - the patient is asked to perform repeated actions of the same type: for example, intensively clenching the fingers into a fist at a fast pace and straightening them. With myotonia, after the first compression of the hands, a tonic spasm of the finger flexors occurs, after which the straightening of the hands occurs slowly and with difficulty. When repeating the same actions, the severity of muscle spasm gradually decreases.
2. Myotonic reaction - a muscle response to mechanical or electrical stimulation, characterized by a prolonged strong contraction and subsequent slow relaxation.
3. Myotonic reflexes - there is a slow relaxation of muscles that contract when causing tendon or skin reflexes.
4. Symptom of muscle roller - when a hammer hits a muscle, in particular the tongue, a patient with myotonia at the site of the blow retains a hole or roller for some time, sometimes a hole surrounded by a roller, which can be observed for several seconds, in severe cases - up to minutes.
5. False Graefe symptom - the patient is asked to look up and then quickly look down. With myotonia, the eyeballs turn downward. And the upper eyelids lag behind, and a strip of sclera remains between the upper eyelid and the edge of the iris, as happens with Graefe's symptom. In the case of myotonia, repeating the same actions leads to a decrease in the lag of the upper eyelid.
6. Symptom of eminence of the thumb - when you hit the eminence of the thumb with a hammer, this finger is adducted, lasting from several seconds to minutes and caused by a spasm of the adductor pollicis muscle.
7. The phenomenon of myotonic spasm of the hand extensors - a blow with a hammer to the hand extensor muscles on the forearm 4-6 cm below the elbow joint causes rapid extension of the hand, followed by its “freezing” for several seconds, and then a slow return to its original position.
8. The phenomenon of squatting - a patient with myotonia always stands on his toes when squatting. If he tries to rest on the entire plantar surface of his feet, while at the same time bringing the medial surfaces of the thighs and shins closer together, he becomes unstable and may fall.
9. Myotonic generalized spasm - a spasm occurs that covers the entire muscle during a sudden sudden movement or when trying to maintain disturbed balance. The patient often falls and is immobilized for some time. Spasm appears in severe forms of myotonia.
It should be noted that diagnosing dysphagia syndrome, which occurs in Kurschmann-Batten-Steinert myotonia, is a difficult task. Differential diagnosis should be made between oropharyngeal and esophageal forms of dysphagia (Table 2).
Laboratory diagnostics
General clinical blood and urine tests for this pathology are not a diagnostic marker. Serum CPK is usually normal or increases several times. A decrease in the concentration of testosterone and IgG in the blood serum is possible. EMG (using needle electrodes) reveals myotonic or pseudomyotonic high-amplitude discharges, increasing in amplitude with muscle contraction and then decreasing. The cycle between identical phases of the muscle's bioelectrical response takes approximately 500 ms. The discharges are accompanied by a characteristic sound phenomenon (“the sound of a dive bomber”).
Given the variety of clinical syndromes, diagnosing Kurschmann-Batten-Steinert myopathy is a challenging task. Here is an observation.
Patient E., 21 years old, upon admission complains of convulsive contractions in the arms and legs, headache, nausea, and increased fatigue.
Born prematurely at up to 32 weeks, he was registered with a pediatric neurologist and was recognized as disabled in childhood, and subsequently as a group III disabled person. In childhood, an MRI of the brain was performed and revealed hypotrophy of the hemisphere cortex of the frontoparietal lobes. Previously, he was examined for perinatal encephalopathy with syncope and liquor discirculation. Was observed for chronic autoimmune thyroiditis. About 1 year ago I began to notice pain in my legs. MRI of the LS section of the spine was performed repeatedly - arthritic changes in the facet joints and osteochondrosis of the discs were revealed. An electroneuromyographic (ENMG) study was carried out repeatedly (in childhood), with the help of which elements characteristic of mitochondrial myopathy were identified. The patient's condition worsened in the last 2 weeks; due to physical activity, the intensity of pain in the leg muscles increased, and convulsive contractions in them intensified, especially when moving.
Upon objective examination, the condition is relatively satisfactory. The skin and visible mucous membranes are pale. In the lungs there is vesicular breathing, no wheezing. The activity of the heart is rhythmic, the tones are muffled. Blood pressure - 110/60 mm Hg. Severe dysplastic physique, hypotrophy of the hands, dysplasia of the small joints of the fingers.
The neurological status includes exophthalmos on the right, Horner's syndrome on the left, nystagmus, and weakness of convergence on both sides. Mild Wurpe symptom. Marinescu's sign - Radovici, more clearly on the right. Tendon reflexes from the limbs are high and equal. There is delayed muscle relaxation after inducing tendon reflexes. The impression of pseudohypertrophy of the thigh muscles. Positive symptom of “muscle roller” in the m.biceps femoralis on both sides, symptom of the extensor pollicis, false Graefe’s symptom. Positive test for myotonia. When lifting the outstretched legs, cramps are noted in the lower third of the thighs. Vibration sensitivity 9–11 seconds. Strumpel's sign on both sides. Muscle weakness in the proximal limbs. S-shaped scoliosis. Limitation of the functional range of motion in the L-spine. Duck-like gait.
Laboratory clinical and biochemical studies of blood and urine revealed no pathology. Blood CPK is normal. ENMG - needle examination of mm.deltoideus, mm.biceps brachii, mm.peroneus longus dextra et sinistra, extensor digiti communis sinistra, deltoideus dextra reveals spontaneous activity, consisting of fibrillation potentials and positive sharp waves, in the form of myotonic, decreasing discharges. Myotonic discharges are accompanied by the characteristic sound of a diving aircraft (Fig. 1).
An MRI of the brain reveals a focal change in the white matter of the frontal region on the right, most likely of vascular origin. Dilation of the ventricles of the brain, cisterns, and intrathecal spaces. MRI signs of decreased blood flow along the left MCA in the M2 segment, and along the right PCA in the P3 segment. ECG - sinus rhythm, bradycardia - 56 beats per minute, signs of diffuse changes in the myocardium. Echocardiography revealed grade 0–1 mitral valve prolapse with minimal regurgitation.
The patient received cardonate, sirdalut, actovegin, cerakson, aevit, ATP-long, glycine.
As a result of the treatment, the patient's condition improved and the frequency of cramps decreased. Mild tetraparesis and amyotrophic syndrome in the arms persist.
Thus, the combination of active and mechanical myoclonic reactions, myopathic syndrome, signs of damage to the cardiovascular system, a progressive course, as well as signs of myotonia on ENMG allowed us to diagnose dystrophic Kurschmann-Batten-Steinert myotonia with mild tetraparesis, frequent cramps, syncope in history, mild impairment of walking function, slowly progressive course. Sop.: consequences of intrapartum damage to the nervous and muscular system in the form of encephalomyelopathy, with liquor-vascular discirculation, atrophic changes, polyglandular insufficiency, mitochondrial disorders.
To make a diagnosis of myotonic dystrophy, it is necessary to take into account the following criteria: autosomal dominant type of inheritance, early onset of the disease, a combination of myotonic and myopathic symptom complexes, multisystem damage, in particular the cardiovascular system (cardiomyopathies, cardiac rhythm and conduction disorders), progressive course. An ENMG study is mandatory. Complex therapy (in combination with sirdalut, glycine) has a therapeutic effect.
Symptoms of the congenital form of Rossolimo-Steinert-Kurshman myotonia
In situations where neurologists diagnose a chronic variant of the disease, the first signs of the presence of an anomaly are pathological processes that arose in the prenatal period. Often, symptoms manifest themselves in a serious decrease in the motor activity of the developing fetus, which is noticed during an ultrasound examination by a doctor in the field of gynecology and obstetrics (during the third trimester of pregnancy). After birth, the child has the following problems:
- Diffuse disorders of muscle tissue - localized at points in the facial, chewing and oculomotor muscle tissues, as well as in the muscles of the distal parts of the limbs.
- Problems with feeding.
- Malfunctions of the breathing apparatus.
- Slow progression of motor skills.
- The appearance of oligophrenia.
Increased child mortality is recorded with increasing symptoms of the disease.
Clinical manifestations
Due to the varying onset of the disease, the clinic distinguishes the following forms according to age:
- congenital form - the manifestation of the disease begins immediately after the birth of the child;
- juvenile variant - the debut of myotonia between the ages of one year and puberty;
- classical form - the onset of clinical manifestations occurs in the second and third decades of life;
- the minimum option is that manifestation occurs late in life—the sixth decade of life.
It is typical that the later the disease manifests itself, the more favorable the course and better the prognosis. The most common form of Steinert disease is the classical one, for which the following clinical symptoms are typical:
- Myotonia - manifested by spasms of the masticatory muscles and flexors of the hands, characterized by atrophic changes in different muscle groups. Myotonic manifestations gradually fade away and muscular dystrophy progresses; outwardly this is expressed in a sad face mask and lack of facial expressions. Dangerous is paresis of the laryngeal muscles with impaired swallowing, as well as weakness of the respiratory muscles, which can result in attacks of respiratory arrest during sleep and the development of pneumonia.
- Cardiovascular disorders - heart rhythm disturbances, hypertrophic changes in the left ventricle detected on ECG, congestive heart failure.
- Endocrine disorders (mainly affecting sexual functions) - reduction in the size of the genital organs, decreased sexual desire, in women - menstrual cycle disorders, obesity.
- General changes of a dystrophic nature are dryness and pigmentation of the skin, partial or complete loss of hair and teeth, early cataracts.
- Central nervous system disorders - fatigue, sleep disorders, apathy, loss of intelligence.
It is especially worth noting the characteristic clinical manifestations of the congenital form of dystrophic myopathy:
- decrease in active movements of the fetus in the womb, detected during ultrasound;
- during the newborn period - lethargy, widespread hypotension, especially in the masticatory, facial, and eyeball muscles;
- preservation and even increase of tendon reflexes;
- feeding problems, breathing disorders such as respiratory distress syndrome;
- delayed physical and neuropsychic development, signs of mental retardation;
- rapid progression of the disease, high risk of sudden death.
Diagnostic options
If you have the above complaints, you should urgently visit a qualified specialist in the field of neurology. To confirm the conclusion, the doctor will prescribe certain clinical procedures, for example:
- Genealogical examination, which will show the presence of an autosomal dominant form of inheritance of the pathology.
- DNA test.
- Electromyographic diagnostics.
- Electroneurographic examination.
- Checking sex hormones.
- Electrocardiographic assessment.
In addition, consultations are held with doctors specializing in problems of hereditary, cardiological, endocrinological, gynecological and other types.
Methods for treating dystrophic myotonia Rossolimo-Steinert-Kurshman
Currently, scientists have not yet been able to draw up a radial plan for getting rid of the symptoms of the described disease. Patients suffering from dystrophic myotonia are prescribed a special diet, which includes a low potassium content in foods. In addition, doctors prescribe the following procedures:
- Do not stay in the cold for a long time.
- Use of quinine, phenytoin and other medications.
- Taking steroid pharmacological drugs with anabolic properties.
- Small dosages of ATP and vitamin complexes are prescribed, in particular B vitamins.