Creutzfeldt-Jakob disease
(CJD) is a rare neurodegenerative disease that has severe effects on the brain. The disease gradually destroys brain cells and causes tiny holes to form in the brain. Patients with CJD have difficulty controlling body movements, have changes in gait and speech, and develop dementia.
There is no cure for this disease. The disease progresses rapidly, and each case leads to death. A person usually dies within 1 year of the onset of symptoms.
Types of Creutzfeldt-Jakob disease
There are different types of Creutzfeldt-Jakob disease. The disease can develop sporadically, without any specific pattern, or it can be inherited.
Sporadic (sCJD
) form develops in people over 60 years of age and occurs in one in a million people every year worldwide.
Another type called familial CJD
, is detected in approximately 5-15% of cases. Inheritance is autosomal dominant, the age of onset of the disease is usually earlier than in sCJD with a longer progression time.
Acquired CJD
- A person can get CJD after eating meat from a cow that has bovine spongiform encephalopathy, commonly called mad cow disease. However, the development of the disease in this way is rare.
Treatment of the abnormal condition
Restoring damaged fibers is currently not available in modern medicine. Doctors can only prescribe symptomatic treatment aimed at relieving mental symptoms, antiepileptic drugs, and medications prescribed for Parkinson's disease. Treatment with known antiviral substances does not achieve any effect, nor does pre-vaccination of patients and livestock. Some effect was detected with the use of Brefeldin A, which stops the proliferation of aggressive proteins in affected neurons, but this effect is temporary. It has not yet been possible to completely get rid of the anomaly, but treatment approaches are under active development.
What is Creutzfeldt-Jakob disease?
CJD
refers to transmissible spongiform encephalopathy (TSE), which destroys the brain over time. CJD is caused by a prion, a misfolded protein that can transfer its shape to healthy variants of the protein. Other types of TSE in humans include Gerstmann-Straussler-Scheinker syndrome and familial insomnia.
Laboratory tests show that transmission can occur between animals, but this does not mean that CJD is transmitted from person to person. However, transmission of infection is possible, for example, through transfusion or transplantation of infected blood or tissue.
Epidemiology
dark green
indicates countries where human cases have been reported;
light green
indicates cases of mad cow disease.
Creutzfeldt-Jakob disease accounts for about 85% of all human prion encephalopathies and affects people of all nationalities and races, men and women, adults and children. There is a slight predominance in the incidence of kuru disease in Aboriginal women of the island of New Guinea, which is associated with the peculiarities of national traditions (ritual cannibalism), when women eat the brains of the dead and receive a high dose of PrPSc. The onset of the disease usually occurs in middle or late age, in typical cases in the fifth decade of life, but can occur at any age. Thus, the age of debut of the classic form varies from 17 to 87 years (the average age is 64 years), while the average age of the new form is much lower and is 29 years.
Creutzfeldt-Jakob disease - symptoms
CJD has a long incubation period and can take decades to manifest itself.
Symptoms appear as the disease destroys brain cells. The person's condition will quickly deteriorate. The symptomatic period lasts on average 4-5 months, and the disease is usually fatal within 1 year.
Characteristic symptoms of CJD are rapid progression of dementia and myoclonus-spasmodic, involuntary movements of muscle groups.
Other common symptoms include:
- changes in mood, personality, or behavior
- memory loss
- impaired judgment
This condition may resemble Alzheimer's dementia or Huntington's disease.
As the disease progresses, coordination and muscle control deteriorate. Over time, a person loses vision, the ability to move and speak. He eventually falls into a coma. Autopsy of brain tissue showed that CJD leads to certain changes that are not found in other forms of dementia. There are several types of Creutzfeldt-Jakob disease. The symptoms and progression of each may vary.
Obvious signs of the disease
Most often, the development of the disease is protracted and gradual, but there are also patients suffering from rapid variation - an acute form, when the disease begins with sudden behavioral changes and damage to cognitive functions. A third of patients complain of conditions such as:
- pain in the head area;
- frequent fainting sensations;
- irritability, depressed mood;
- absentmindedness;
- intermittency of night sleep, insomnia;
- decreased memory level;
- partial loss of sound perception;
- apathy to what is happening around, loss of activity;
- lack of sexual desire;
- transformation of behavioral reactions;
- sometimes there is causeless euphoria or panic attack, delusions or hallucinations;
- partial loss of coordination;
- forgetting words, inability to perform simple mathematical calculations in the mind.
With the further course of the disease, subsequent degenerative symptoms develop:
- periodic paralysis of the muscular system;
- epileptic seizures;
- severe trembling in the limbs;
- nervous twitching of individual muscle groups, most often the nasolabial triangle and eyelids;
- progressive dementia, loss of thinking abilities;
- speech dysfunction up to the complete collapse of conscious sound production;
- The patient loses sensitivity to external influences.
At the final stage of the disease, the following is observed:
- profound dementia;
- complete detachment, lack of contact;
- loss of control of excretory functions, the patient requires constant supervision and care;
- atrophy of the muscular system, degeneration of unconditioned reflexes (for example, swallowing);
- falling into a coma with subsequent death.
Creutzfeldt-Jakob disease - causes
Scientists believe that prions are responsible for Creutzfeldt-Jakob disease. Creutzfeldt-Jakob disease
has a long incubation period and is difficult to diagnose. They do not contain genetic information in the form of nucleic acids such as DNA or RNA.
CJD can be inherited or acquired. In some cases, it develops sporadically, without a specific pattern.
Sporadic CJD
In 85% of cases the disease is sporadic. This means that there is no clear reason why it develops.
Familial Creutzfeldt-Jakob disease
A person can have familial CJD—10% to 15% of cases are inherited. The disease develops when there is a change in the gene that controls the formation of prion proteins. Prions contain no genetic information and do not require genes to reproduce. However, a genetic mutation can cause prions to function incorrectly.
Scientists have identified several mutations in the gene. A specific mutation can affect how often the disease runs in a family and which symptoms are most noticeable. However, not everyone who has mutations in the prion gene develops CJD.
Prognosis and prevention
Since it is impossible to stop the manifestations of the disease using medical methods, and the transformation of the prion occurs at a high speed, the only outcome for sick patients, unfortunately, is death. The average life expectancy after the first signs appear is 8-12 months in 90% of cases. Only a small proportion of patients continue to live for 24-26 months.
Preventive measures include careful processing of medical instruments before surgery, analysis of tissue intended for transplantation purposes and avoidance of meat products from cattle. The most frequent cases of incidence of the described pathology are recorded in countries such as Great Britain, Chile, Slovakia and Israel.
Acquired Creutzfeldt-Jakob disease
There is no evidence that CJD can be transmitted from one person to another through casual contact. However, transmission of infection can occur during procedures involving damage to brain or nervous system tissue. These procedures may include:
- corneal transplant
- implantation of electrodes
- meningeal graft, outer membrane graft
Less than 1% of CJD cases are acquired. Transmission of CJD can occur by eating meat from a cow that has had bovine spongiform encephalopathy. A person cannot acquire CJD through food.
Changes occurring in the body
Pathological protein, multiplying and accumulating in the body, leads to:
- the development of spongiform degeneration of nerve cells, which consists of the formation of a large number of intracellular vesicles and expansion of neuron processes. These changes create a “spongy” fine-mesh effect during microscopic examination of a brain biopsy. In some cases, it cannot be detected using available research methods;
- replacement of microglia with connective tissue;
- a significant decrease in the number of neurons in the cerebral cortex, subcortical formations (basal ganglia, thalamus), cerebellum and brainstem structures;
- amyloid accumulation in 10-15% of cases.
Creutzfeldt-Jakob disease - clinic
No test can confirm the diagnosis of CJD. Only a brain biopsy can do this, but this procedure is too risky. However, certain tests and imaging techniques can help your doctor make a diagnosis.
During the examination, the doctor can detect muscle spasms and test the person's reflexes. They may be more active than usual. Additionally, muscles may be overly toned or weak, depending on where the disease affects the brain.
An eye test may show vision loss, and an EEG may reveal unusual electrical impulses that may characterize the disease.
An MRI scan will help rule out stroke as the cause of your symptoms. An MRI scan may also show changes consistent with CJD.
Your doctor may test your spinal fluid using a spinal tap to rule out other causes of dementia. This test can show whether there is an infection or high blood pressure in the central nervous system. If the 14-3-3 protein is present in the cerebrospinal fluid and a person has typical symptoms, it may indicate CJD.
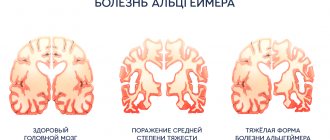
A brain biopsy after death shows that the brain tissue is spongy, with tiny holes where clumps of nerve cells have been destroyed.
NEUROLOGY
CREUTZFELDT-JACOB DISEASE
O. Habib O. Habib
G
Serpentine encephalopathies are infectious degenerative diseases.
Some family forms are genetically determined. In brain tissue, pathological protein particles are found - prions (PrPres), which are a stable pathological (devoid of nucleic acid) copy of the prion protein (PrP), present in the healthy nervous tissue of all mammals. PrP is a membrane sialoglycoprotein consisting of 253 amino acids; it has a two-dimensional spatial structure and is encoded by a single gene on the short arm of chromosome 20. The pathological form of the protein is found only in brain tissue affected by infectious spongiform encephalopathy (ISE). PrPres and PrP have different spatial structures and properties: PrPres is insoluble in water, insensitive to proteases, has a larger number of b-chains and a smaller number of a-helices in its structure. It is believed that the conversion of PrP to PrPres occurs during transcription. It is not yet clear whether PrPres is both pathogenic and virulent (since no nucleic acid was detected, but in the experiment it was possible to obtain PrPres from PrP in the acellular system) or whether PrPres contains a certain virus (i.e., there is a nucleic acid), which causes infectious transmission, and PrPres only produces a toxic effect. The existence of a particle with an independent genome best explains the presence of several PrPres species. One hypothesis suggests the existence of an as yet unrecognized molecule, which, covering the protein and the virus, prevents the nucleic acid from being “seen.” Genetic predisposition.
The gene responsible for PrP synthesis is characterized by polymorphism.
Codon 129 can code for either methionine or valine. 50% of the population are heterozygous for codon 129 (methionine/valine), 40% are homozygous for methionine and 10% are homozygous for valine. And since all patients with IGE are homozygotes, the loss of polymorphism can be considered a factor predisposing to the onset of the disease. Varieties of PrPres.
In sheep affected by IGE, up to 20 varieties of PrPres are verified and, accordingly, different incubation periods and the nature of the CNS lesion are observed.
Only one type of PrPres is typical for IGE in cows. For Creutzfeldt-Jakob disease (CJD), 4 varieties of PrPres are known (by immunoblotting). Types of CJD.
CJD is a type of human IGE.
In addition to CJD, this group of diseases includes Kuru disease (a “laughing” disease, endemic to New Guinea, characterized by 100% mortality), Gertsmann Straussler Scheinker disease and fatal familial insomnia. Based on the clinical picture and mode of transmission, the following types are distinguished: classical/sporadic, familial/genetic, iatrogenic and British IGE. The classic form
is described by Creutzfeldt (1920) and Jacob (1921).
The incidence in the world is 0.5-1 case per 1 million people per year; in France, 30-50 cases are recorded per year; 70% of all cases of CJD occur in the classic form. The disease usually debuts at 55-75 years of age; in the 70 years since its description, 20 exceptions have been registered - juvenile cases (16-40 years), which are now considered to be the British variant. Characteristic is a gradual progression of symptoms of central nervous system damage; in 13% of cases, the disease “unfolds” within a few days. Clinical picture:
initial symptoms in 35% of cases are depression and disorder of mental functions, in 34% - neurological symptoms with predominant damage to visual functions and the cerebellum;
in 21% - combined. Symptoms progress rapidly: profound dementia develops, often in combination with mutism. Ataxia, tremor and rigidity cause locomotor disorders up to and including immobilization. Mixed cortico-retinal blindness occurs; at a later stage, pyramidal disorders, myoclonus and epileptic seizures are common. Death occurs before a year has passed from the onset of the first symptoms, in 70% of cases by the 5th month. Paraclinical data are not specific: the EEG shows periodic convulsive discharges, the cerebrospinal fluid is normal (PrPres has not yet been determined), the computed tomogram shows atrophy of the cerebral and cerebellar cortex. Pathomorphologically
they determine a decrease in the number of cortical neurons, vacuolization of brain tissue, which takes on the appearance of a sponge;
glial, mainly astrocytic, proliferation. There are no signs of inflammation. PrPres are verified using immunohistochemical analysis and immunoblotting. Since 1968, brain tissue extract has been inoculated into susceptible animals (primates, rodents, cats). The diagnosis is made on the basis of clinical manifestations and is confirmed by pathological data and infection of laboratory animals. Epidemiology.
Despite the abundance of data, it has not yet been possible to explain the origin of the classical form.
Some time ago, nutritional contamination was discussed due to the increasing incidence in Iceland. The discussion resumed after the verification of IGE in cows in 1986. Affected people are characterized by increased trauma and a history of frequent surgical interventions. It is possible that representatives of certain professions - shepherds, farmers, doctors - are at increased risk of the disease. The familial (genetic) form
accounts for 5-6% of all cases of CJD and is inherited by an autosomal dominant mechanism.
Most often, this mutation is the replacement of aspartate acid with asparagine at position 178 of the PrP molecule. The disease debuts 5-10 years earlier than in the classical form, and the course of the disease is the same. There are isolated ethnic groups where the incidence of CJD is 30-100 times higher than for the general population, which is due to the characteristics of the genotype and the specificity of mutations. These are Libyan Jews (Middle East and Mediterranean), Slovak and Chilean communities, in which intra-family marriages are common. Iatrogenic CJD
has appeared in recent years, there are few cases, but their drama in psychological, ethical and medico-legal aspects is obvious.
Based on the type of infection, two groups are distinguished. Central infection occurs during operations, surgical procedures, through grafts and surgical instruments. There are 24 known cases of infection during transplantation of dura mater taken from victims of CJD, 1 case during corneal transplantation, 2 cases after stereotactic surgery. The incubation period after inoculation is from 7 to 120 months. It is logical to conclude that the “infectious principle” (virus?) is very resistant to the effects of traditional antiseptics, which are used to treat instruments and transplants. Therefore, they must be reliably disinfected, and the best solution is to use disposable instruments. Known cases of peripheral infection are associated with the administration of GH. Of all 50 registered, 70-80% are in France. 4 cases in Australia involved the administration of pituitary TG to induce ovulation. Today it is obvious that replacing natural GH with recombinant (genetic engineering product) will completely eliminate the risk. The latent period is 18-28 years. The clinical picture is more similar to the symptoms of Kuru disease - neurological symptoms prevail over intellectual degradation. In most patients, loss of polymorphism at codon 129 and characteristic prion “behavior” are observed when performing immunoblotting. The experiment proved the possibility of infecting animals with the blood of patients. Thus, the safety of donor tissues and organs must be ensured, and their use must be justified. British version.
Between 1995 and 1997, there were 14 cases of CJD in young people in the UK (and another in France). Common to all cases were: normal PrP gene, homozygosity at codon 129, absence of any risk factors, spatiotemporal coincidence (incubation for several years), characteristic “behavior” of PrPres during immunoblotting, reminiscent of that in IGE cats infected through nutrition . A hypothesis has emerged about the possibility of nutritional contamination through consumption of meat from sick animals. The situation will become clearer in the next two years with the completion of experiments on transgenetic mice. It is possible that currently unknown protective factors may be discovered.
Literature:
Beauvais P. La maladie de Creutzfeldt-Jakob. La plus important des maladies a prion. La Presse Medical 1997;26:3787-92.
Risk of developing tension-type headaches among immediate family members and spouses of people with this problem
E. Nurmukhametova Ye. Nurmukhametova
G
Almost everyone has experienced tension-type pain (TPN), a syndrome characterized by stiffness of the neck and neck muscles with a sensation of pain in the back of the head. This is a fairly rare occurrence for most people, but approximately 3% of the population (5% of women and 2% of men) suffer from frequent attacks (called chronic TTH). The etiology of this disease remains unknown. Some authors point to a connection between tension-type headache and psychoneurosis. Genetic factors also cannot be excluded, since in many cases there is a family history of chronic tension-type headache.
| Risk of developing TTH | Number of first-degree relatives suffering from HDN | Population relative risk, estimated N/R (95% confidence interval) | |
| Within 1 year: spouse's immediate family | observed (N) | expected (O) | 3,18 (2,26 — 4,31) 1,23 (0,26 — 3,49) |
| 36 | 11,31 | ||
| 3 | 2,43 | ||
| During life: spouse's immediate family | 71 | 22,61 | 3,14 (2,50 — 3,86) |
| 4 | 4,85 | 0,82 (0,23 — 2,68) | |
The aim of this study was to determine the prevalence of TTH among immediate family members and spouses of persons suffering from this disease. 122 patients with symptoms characteristic of tension-type headache were examined. Spouses and immediate family members were interviewed by telephone. Adjusted for gender, age and the average incidence of the disease among the population (3% per year and 2 times more often throughout life), the results presented in the table were obtained. Thus,
immediate relatives of patients suffering from tension-type headache have an increased risk of developing this pathology (3 times higher than the population risk), which confirms the important role of genetic factors in the etiology of the disease.
Literature:
Ostergaard S, Russel MB, Bendstsen L, et al. Comparison of index degree relatives and spouses of people with chronic tension headache. BMJ 1997; 314:1092-3.
Creutzfeldt-Jakob disease - treatment
There is no specific treatment for CJD, and no medications can control the disease or slow its progression. Doctors strive to relieve symptoms and eliminate discomfort.
Opioid medications may help relieve pain. In addition, clonazepam and sodium valproate will reduce involuntary movements such as muscle twitching.
In later stages, it is necessary to turn the person over more often to prevent bedsores. The patient is given nutrients intravenously.
Etiology, pathogenesis and routes of infection
See also: Prion disease
Once in the body, the prion settles on the surface of the cell, interacting with normal proteins and changing their structure to pathological.
Pathological proteins accumulating on the cell surface block processes occurring on the membrane, which leads to cell death.
Proteins accumulating on the cell surface trigger apoptosis (the programmed process of cell death).
The cell, trying to get rid of proteins on the surface, begins to produce reactive oxygen compounds, but the protein on the surface prevents them from leaving the cell. Active substances destroy the cell itself.
Inflammatory processes begin around the affected cells with the participation of highly active enzymes that affect neighboring healthy cells.