Epilepsy is a serious chronic disease characterized by uncontrolled spontaneous seizures of various types. The disease may be congenital or acquired.
The first epileptic seizures can occur at any age; their development is due to many reasons: from genetic predisposition to previous neuroinfections and traumatic brain injuries.
There are certain provoking factors, the presence of which can increase the likelihood of epileptic seizures. If they are present, the patient needs to regularly visit a neurologist to prevent attacks and receive timely medical support.
Causes of the disease
Epilepsy can be idiopathic or symptomatic in nature. The reasons for the development of idiopathic epilepsy remain unclear. The pathology can be diagnosed in people both at an early and mature age, but is most often found in children. Scientists believe that one of the most likely causes of idiopathic epilepsy is hereditary predisposition.
The occurrence of symptomatic epilepsy is associated with one or another negative circumstance that has a negative impact on the structures of the brain. Thus, an epileptic attack is provoked by the following factors:
- repeated flashes of light and color;
- repetitive sounds;
- bright changing pictures, video effects;
- poisoning of various kinds;
- consumption of alcoholic beverages, drugs;
- taking certain medications;
- oxygen starvation;
- hypoglycemic attack - when there is a sharp drop in blood sugar levels.
You need to know that an epileptic seizure in a healthy person can be caused by one of the last three reasons.
Types of childhood epilepsy
The following types of seizures are distinguished in children:
- Absence or non-convulsive generalized. A common type, manifests itself from birth to puberty. Difficult to detect, absence lasts 5-20 seconds. The child “switches off” from the world around him. Such attacks may be accompanied by a slight tilting of the head and trembling of the eyelids. With equal probability, after puberty they may disappear or, on the contrary, intensify.
- Absence - rolling of the eyes and trembling of the eyelids
- Infantile spasms appear at 2-3 years of age. Early in the morning after waking up, the body is not completely controlled for several seconds. The baby nods his head, straightens his legs and arms, brings them to his chest, and his torso leans forward.
- Atonic is an externally short-term fainting spell, during which the body completely relaxes and consciousness becomes darkened for a few seconds.
- Full-blown seizures with severe convulsions, loss of orientation in space.
- Night attacks that occur during sleep can be recognized by sleepwalking. As a rule, they go away by the period of growing up on their own without the necessary treatment. Symptoms of childhood epilepsy are often mild, and parents and relatives do not notice the presence of the disease for a long period. The similarity of manifestations with other diseases complicates the diagnosis and prescription of correct treatment. The average age of identification of patients is 5-18 years.
It is possible to determine with certainty whether it is possible to cure epilepsy completely in a teenager or young children or not, and how to do this, only in some of its variants. Those whose causes of development have been established in detail. More often we have to hope for positive dynamics, with the weakening of convulsions and a decrease in seizures.
Causes of epilepsy in adolescents
Epilepsy in adolescence is most often a continuation of a disease that began in childhood. Epileptic seizures in adolescents can either increase or decrease. Puberty is associated with global changes in the body, preparation for growing up and puberty. Therefore, it is quite difficult to predict how epilepsy will manifest itself.
The appearance of symptomatic (secondary) epilepsy in adolescence is usually caused by the following negative factors:
- infectious disease of the brain (meningitis, encephalitis);
- traumatic brain injury;
- severe intoxication with alcohol, drugs or chemicals;
- thyroid diseases;
- metabolic disorders.
Epilepsy manifests itself in specific seizures that occur spontaneously and cannot be controlled. If more than one attack occurs, you must immediately consult a neurologist.
Make an appointment
How to make an appointment with a neurologist
To make an appointment with a neurologist at a time convenient for you, use the online form or call the clinic’s contact number. Children experiencing epilepsy for the first time may require hospitalization to undergo a full examination. We are located in the Central Administrative District, close to the Novoslobodskaya, Tverskaya, Chekhovskaya, Belorusskaya and Mayakovskaya metro stations.
Also, in case of attacks that do not go away for more than 5 minutes or recur after a short period of time, you can call an ambulance.
Provoking factors in children
Epilepsy is not considered a childhood disease, but is more often diagnosed at an early age. Depending on the form of the disease, it can manifest itself in different ways. Epilepsy is accompanied by convulsions and loss of consciousness.
The most common causes of epilepsy in children are:
- neck and head injuries during birth;
- hypoxia (oxygen starvation) of the fetus;
- infectious diseases suffered by the mother during pregnancy;
- maternal abuse of alcohol and drugs during pregnancy.
The disease can appear in a child immediately after birth or several years later. Minor convulsive movements in children under three months of age may be a consequence of an immature nervous system. However, if they are present, you should consult a neurologist to exclude pathology. If seizures continue to appear after three months of life, intensify and occur spontaneously, it is necessary to show the child to a pediatric neurologist.
Parents need to pay attention to the following symptoms in their child:
- fading;
- loss of concentration;
- no reaction to stimuli;
- high temperature;
- fainting;
- lethargy;
- headache;
- nausea;
- stomach ache;
- dizziness.
If these symptoms occur, call a doctor immediately or take your child to the hospital.
How to determine epilepsy in a child
The most common and recognizable symptom is seizures accompanied by loss of consciousness. It is worth noting that this symptom is characteristic of a severe form of the disease.
The dangerous thing about the disease is the fact that the attack occurs suddenly and unpredictably. Often patients are forced to carry medications with them to stop an attack, otherwise death may occur.
Doctors distinguish more than 50 subtypes of epilepsy, each of which has its own characteristic features and manifestations. For a person not involved in medicine, it may be difficult to recognize this disease. Therefore, parents should carefully monitor the child’s health and promptly respond to any changes in his behavior.
In infants and one-year-old children, the disease manifests itself in the same form. You should immediately contact an epileptologist if you experience the following symptoms:
- temporary numbness of the facial muscles, and then their rapid contraction, the face for some time turns into a motionless mask;
- when feeding a baby, the space between his upper lip and nose turns blue;
- the child fixes his gaze on one point for a long time;
- The baby's limbs twitch involuntarily.
The older the child becomes, the more pronounced the symptoms of the disease. The nature of the symptoms of epilepsy at the ages of 3 and 8 years has significant differences. In addition to the above-described signs of the disease, you should pay attention to the behavior and character of the child. Children who are most likely to have epilepsy are aggressive and irritable, often restless. The child finds it difficult to study at school and is unable to build relationships with peers.
Provoking factors in adults
Hereditary forms of epilepsy do not always appear at an early age. There are cases where patients of older age groups experienced their first epileptic attack, and upon examination it turned out that there was a genetic predisposition.
The occurrence of epileptic seizures in adulthood is provoked by other factors:
- suffered a stroke;
- brain tumors;
- multiple sclerosis;
- metabolic disorders;
- infectious diseases with brain damage (meningitis, encephalitis, etc.);
- alcohol, drugs and other intoxications.
Traumatic brain injuries can cause epilepsy in people of any age. Damage to the bones of the skull, hemorrhage in the brain and disruption of the integrity of soft tissues may be accompanied by the development of pathological processes with secondary epilepsy.
Often the attack is accompanied by dislocations and fractures. Seizures can also be vegetative - without loss of consciousness or convulsions. They are characterized by the following types of symptoms:
- increased heart rate;
- excessive sweating;
- flatulence and cramping abdominal pain.
Nonconvulsive epilepsy can be caused by factors such as predisposition and exogenous influences. Epilepsy in adults is treatable. Only an accurate diagnosis in the early stages can guarantee success.
Variants of the disease
Epilepsy in adolescents can occur in different ways. This is due to the fact that epileptic foci can be located in different parts of the cerebral cortex. Moreover, the form of the disease does not depend in any way on its causes, and over time it can become milder or more severe.
The main forms of epilepsy:
- myoclonic (Jantz syndrome). Develops against the background of puberty. At an early stage, twitching of the muscles of the arms and shoulder girdle is noted;
- absence The mildest form of epilepsy, in which seizures occur without loss of consciousness or memory. But without treatment it can develop into a generalized form. Attacks occur from once a day to once every couple of weeks;
- with generalized attacks. The most severe form of the disease. Accompanied by attacks with a frequency of 1 time per week to 1 time per year. Sometimes absence seizures or myoclonic seizures may occur;
- catamenial. A subtype of epilepsy that occurs against the background of hormonal changes associated with the formation of the menstrual cycle. After its normalization, epilepsy usually goes away.
The first signs of epilepsy
Often, the patient and his family are not aware of the presence of epilepsy until the first attack. It can be quite difficult to determine the reasons for its development and the specific irritant for the first time, however, the fact that an epilepsy attack is approaching can be suspected if the patient has certain signs:
- headache a few days before an epileptic seizure;
- sleep disorders;
- severe stress;
- nervousness and irritability;
- decreased appetite;
- loss of appetite.
During a convulsive attack, muscle tension and lack of response to any irritants are noted - patients do not hear sounds, do not respond to touch, pain, their pupils do not narrow or dilate. After an epileptic seizure, people become lethargic and drowsy; to restore strength and normalize their condition, they need rest and good sleep.
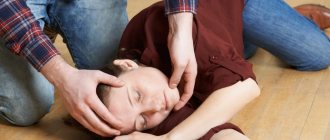
First aid during a seizure
During an epileptic attack, it is important not to get confused and take all necessary measures to prevent injury. The child must be placed on a flat, not hard surface. To prevent your baby from choking on saliva, be sure to lay him on his side. If the mouth opens during a seizure, a handkerchief should be placed between the teeth without restricting the child's breathing.
It is strictly forbidden to forcefully open your mouth or hold your tongue. Also, you should not give medication to the patient during a seizure. The attack usually lasts no more than 5 minutes. After the seizure stops, it is necessary to check the baby’s breathing, and, if it is absent, perform artificial respiration. Be sure to wait for the child to come to his senses, do not leave him alone.
If the seizure occurred for the first time or lasted more than 5 minutes, or if the child was injured during the seizure, it is recommended to call an ambulance.
Epilepsy in adolescents: symptoms
Epilepsy is characterized by a disturbance in the activity of cells in the brain, when the excitation of neurons prevails over the processes of inhibition. As a result, epileptic seizures occur, which are very diverse in their manifestation. In adolescents, focal seizures are most common, possibly with secondary generalization, but generalized seizures are not excluded.
Focal epileptic seizures occur in a specific area of the brain and affect neighboring tissues. The clinical picture of an epileptic seizure in a teenager will depend on the area where the pathological arousal originates:
- Temple area. Epilepsy most often affects this part of the brain. During an epileptic attack, the patient may experience strange, uncontrollable feelings and emotions. Increased anxiety occurs;
- frontal region. Patients develop muscle weakness and speech impairment. Epileptic seizures often occur during sleep, which is accompanied by turns of the head, chaotic movements of the legs;
- parietal region. Patients experience sensory attacks: a person may feel tingling in the limbs, warmth or cold, numbness in some parts of the body;
- occipital region. An epileptic attack disrupts the functioning of the visual organs. The patient's eyeballs begin to move from side to side and the eyelids twitch. Flashes of light, patterns, images appear before your eyes. An epileptic attack is often accompanied by pain. Before and after the attack, the patient may have a severe headache.
During a generalized attack, pathological arousal affects the entire brain. This type of epilepsy significantly impairs the quality of life, as it is associated with the risk of injury to the head or face during an attack. Generalized epilepsy is usually accompanied by an aura - subjective sensations that predict an attack. Among generalized attacks there are:
- tonic-clonic. The attack begins with a tonic phase, when the whole body tenses sharply and an involuntary cry can escape. Next, the patient experiences convulsive twitching. During an attack, a person may lose consciousness. An epileptic attack usually does not last long (up to five minutes), after which the person feels very tired or may fall asleep. Tonic-clonic seizures, in addition to the main problems, cause significant psychological discomfort to the teenager. Anxiety increases because it is impossible to predict when the next attack will occur. Self-doubt develops, and it is difficult for the teenager to socialize.
- absence seizures. A characteristic feature of this type of attack is a loss of consciousness for several seconds or minutes. The person freezes and does not respond to external stimuli. Absence seizures may be accompanied by involuntary twitching of the fingers and facial muscles. Absence seizures create difficulties for learning in a group, since during an attack the teenager misses some of the information presented by the teacher. The attack occurs suddenly, sometimes without an aura. In this case, a person can interrupt mid-sentence, and after the attack ends, continue the conversation.
- atonic attacks. In this case, the muscles sharply lose tone and the person falls. There is a high chance of being seriously injured during an attack.
The nature of epileptic seizures usually remains stereotypical. As the disease progresses without adequate therapy, attacks will become more frequent and their intensity will increase.
Epilepsy in childhood
Part 4. Read the beginning of the article in No. 6 and 10, 2014
Epilepsy in preschool children (4–6 years old)
Preschool children are characterized by the onset of idiopathic partial epilepsy with frontal paroxysms, benign occipital epilepsy with an early onset (Panagiotopoulos syndrome), as well as Landau-Kleffner syndrome.
Idiopathic partial epilepsy with frontal paroxysms
The disease was first described by A. Beaumanoir and A. Nahory (1983). The age of patients at the onset of epilepsy is 2–8 years. This type of epilepsy accounts for about 11% of cases of all idiopathic focal epilepsies. The disease manifests itself in the form of several types of seizures: daytime (complex partial, motor automatisms, sometimes absence-like) and nighttime (hemifacial motor seizures, versive, sometimes with “post-crisis” deficit and/or secondary generalized seizures). The frequency of attacks varies from 1 episode per month to 1 attack per few weeks (the duration of the active period of the disease ranges from 1 year to 6 years). EEG data are quite heterogeneous and do not have a single specific pattern (in some patients, EEG changes are comparable to those in benign childhood epilepsy with centrotemporal peaks; others have only focal slow activity; intermittent frontal discharges are recorded in the ictal period). The prognosis of the disease is quite favorable (spontaneous remission). A transient decrease in cognitive functions (short-term memory, operational functions, etc.) is observed during the active period of the disease; then they gradually recover [3–6].
Benign occipital epilepsy with early onset (Panagiotopoulos syndrome)
One of the benign occipital epilepsies of childhood. The disease debuts between the ages of 1 and 14 years (peak incidence at 4–5 years of age); occurs approximately 2 times more often than the Gastaut variant. Autonomous nocturnal attacks are characteristic; Due to autonomic symptoms and nausea, seizures are poorly recognized. In the early stages of the disease, eye deviation and behavioral disorders are observed (in 50% of cases, attacks can become convulsive in nature). The duration of attacks is 5–10 minutes; in 35–50% of patients they progress to autonomic focal status epilepticus (sometimes with secondary generalization). EEG records peaks or paroxysmal discharges. Two thirds of patients have at least one EEG study with signs of occipital paroxysms (most often occipital peaks); the remaining third of patients have only extra-occipital peaks or short generalized discharges. In approximately 33% of cases in children with Panayiotopoulos syndrome, multifocal peaks in two or more cerebral areas are recorded on the EEG (single peak foci are considered rare). The prognosis for Panayiotopoulos syndrome in terms of long-term remission and cognitive functions is relatively favorable. Treatment of early-onset benign occipital epilepsy is predominantly aimed at controlling seizures in the acute phase (diazepam) [3–6].
Landau–Kleffner syndrome (acquired epileptic aphasia)
The overwhelming majority of cases of the disease occur at the age of 4–5 years, although the disease (acquired aphasia and epileptiform discharges in the temporal/parietal areas of the brain) can debut earlier - in the second or third year of life. The cause of this relatively rare condition is unknown. Landau–Kleffner syndrome is characterized by loss of speech skills (impressive and/or expressive aphasia). These speech disorders, as well as auditory agnosia, appear in children who have not previously had deviations in psychomotor and speech development. Concomitant epileptic seizures (focal or generalized tonic-clonic seizures, atypical absences, partial complex, rarely myoclonic) are recorded in 70% of patients. Some children have behavioral disorders. Otherwise, the patients’ neurological status usually has no significant disturbances. A specific EEG pattern is not typical for Landau–Kleffner syndrome; epileptiform discharges are recorded in the form of repeated peaks, sharp waves and peak-wave activity in the temporal and parieto-occipital regions of the brain. Epileptiform changes in sleep state are intensified or observed exclusively during sleep. Although the prognosis of Landau–Kleffner syndrome is relatively favorable, the onset of the disease before the age of 2 years is always associated with an unfavorable outcome in acquiring and/or restoring speech communication skills [3–5, 9].
Epilepsy in school-age children and adolescents
Upon reaching school age, children and adolescents are at risk of exposure to a whole group of age-dependent epilepsies, among which the most relevant are childhood absence, benign with centrotemporal peaks, benign occipital (Gastaut variant), juvenile absence, juvenile myoclonic (Jantz syndrome) and a number of other forms of the disease.
Pediatric absence epilepsy (pycnolepsy)
The peak onset of the disease occurs in early school age (about 7 years), although childhood absence epilepsy can debut from 2 to 12 years. It rarely debuts before the age of 3. Refers to idiopathic forms of the disease; all cases are considered genetically determined (autosomal dominant inheritance with incomplete penetrance). Childhood absence epilepsy is characterized by frequent repeated absence attacks (up to several hundred per day). In this case, absence seizures are the only or leading type of epileptic seizures; the likelihood of generalized tonic-clonic seizures is significantly lower than with juvenile absence epilepsy. Diagnosis of childhood absence epilepsy is carried out based on clinical manifestations and EEG data (a typical EEG pattern is bursts of generalized high-amplitude peak-wave activity with a frequency of 3 per 1 sec, suddenly appearing and gradually stopping). The prognosis for childhood absence epilepsy is relatively favorable [3, 5, 6].
Benign epilepsy of childhood with centrotemporal peaks (rolandic epilepsy)
Described by P. Nayrac and M. Beaussart (1958). In the age group 0–15 years, it occurs with a frequency of 5–21 per 100 thousand (8–23% of all cases of epilepsy), being the most common form of idiopathic epilepsy in children. The age of children at the time of the debut of rolandic epilepsy varies from 3 to 14 years (peak - 5–8 years). Cases of the disease in patients under 2 years of age are extremely rare. The disease manifests itself in the form of nocturnal tonic-clonic seizures with a partial (focal) onset, as well as daytime simple partial ones (emanating from the lower cortical regions - the region of the central Rolandic sulcus). The frequency of attacks is usually low. Rolandic epilepsy is characterized by a specific somatosensory aura (pathological sensations in the buccal-oral area), as well as hypersalivation, speech arrest, tonic-clonic or tonic convulsions of the facial muscles. The patient does not lose consciousness during the attack. EEG data is characterized by the presence of peak-wave complexes localized in the central temporal regions. In the interictal period, patients' EEG records specific complexes in the form of high-amplitude 2-phase peaks accompanied by a slow wave. Rolandic peaks are localized in one or both hemispheres (isolated or in groups: in the middle temporal - T3, T4, or central - C3, C4 areas). By the time the disease debuts, the psychomotor development of children is normal. Subsequently, the disease is characterized by an almost complete absence of neurological and intellectual deficits. Many patients go into remission during adolescence; A small proportion of children have impairments in cognitive functions (verbal memory), as well as various speech disorders and decreased academic performance (at school) [3–6, 9].
Benign occipital epilepsy with late onset (Gastaut variant)
Focal form of idiopathic epilepsy of childhood (Gastaut syndrome) with a later onset than Panayiotopoulos syndrome. The disease onsets between ages 3 and 15 years, but peaks at approximately 8 years of age. Characterized by short-duration attacks with visual disturbances (simple and complex visual hallucinations), complete/partial loss of vision and illusions, deviation of the eyes, followed by clonic convulsions involving one side of the body. Up to 50% of patients experience migraine or migraine-like cephalgia after the attack. EEG data resemble those in Panayiotopoulos syndrome: in the interictal period, normal main activity of the background recording is recorded in combination with epileptiform unilateral or bilateral discharges in the occipital leads in the form of peak-wave complexes (high-amplitude biphasic peaks with a main negative phase followed by a short positive phase) in combination with negative slow-wave activity. When the patient opens his eyes, epileptiform activity disappears, but resumes again 1–20 seconds after closing the eyes. The prognosis for benign occipital epilepsy with late onset (Gastaut variant) is relatively favorable, but, due to the possible pharmacoresistance of the disease, it is ambiguous [3–5, 9].
Juvenile (youthful) absence epilepsy
The juvenile variant of absence epilepsy belongs to the idiopathic generalized forms of the disease. Unlike childhood absence epilepsy, the disease usually debuts at puberty, pre- or post-puberty (9–21 years, usually 12–13 years). The disease manifests itself as typical absence seizures, myoclonus, or generalized tonic-clonic seizures. The likelihood of a debut in the form of generalized tonic-clonic seizures in juvenile absence epilepsy is slightly higher (41% of cases) than in childhood absence epilepsy. The EEG pattern characteristic of this type of epilepsy has the form of peak-wave activity with a frequency of 3 Hz - symmetrical and bilaterally synchronized. Polypeak-wave activity, which occurs in some patients, should be alarming in terms of transformation of the disease into juvenile myoclonus epilepsy. The prognosis is relatively favorable - there is a high probability of remission in late adolescence [3–6, 9].
Juvenile (youthful) myoclonus epilepsy (Jantz syndrome)
The disease was described by D. Janz and W. Christian (1957) as one of the subtypes of idiopathic generalized epilepsy (another name: “impulsive petit mal”). Usually debuts at the age of 8–26 years (usually at 12–18 years). The hallmark of the disease is myoclonic seizures. Isolated myoclonic twitching in the upper extremities is characteristic, especially soon after awakening. Most children have generalized tonic-clonic seizures, and about a third of patients have absence seizures. Seizures are often triggered by sleep deprivation. Myoclonic seizures are accompanied by short bursts of generalized spike-wave or polyspike-wave complexes during EEG [3–5, 9].
Familial temporal lobe epilepsy
This genetically heterogeneous syndrome is characterized by relatively benign simple or complex partial (focal) seizures with a pronounced mental or autonomic aura. The disease usually debuts in the second (about 11 years) or early third decade of life (more often in adult individuals). Usually occurs against the background of normal development of the central nervous system. MRI of the brain does not reveal any pathological structural changes in the hippocampus or temporal lobes. EEG data allows recording epileptiform activity in the medial and/or lateral areas of the temporal lobes. Seizures in familial temporal lobe epilepsy are often easily controlled medically with traditional antiepileptic drugs [3–6, 9].
Mesial temporal epilepsy
It often debuts in adolescents and is manifested by limbic seizures. In typical cases, in patients with a history of febrile convulsions, temporal lobe seizures occur after a seizure-free interval, which at first respond well to drug control. Subsequently, in adolescence or upon reaching adulthood, relapses of the disease are observed. MRI of the brain may show hippocampal sclerosis in patients, which is considered a key feature of this form of epileptic syndrome. All limbic seizures are more or less refractory to pharmacotherapy [3, 5].
Familial mesial temporal epilepsy
This genetically determined heterogeneous epileptic syndrome was described by P. Hedera et al. (2007). The disease debuts at different ages, but most often in the second decade of life. In contrast to the mesiotemporal epilepsy described above, children usually have no history of febrile seizures. In most cases, patients experience simple focal seizures with the appearance of déjà vu, periodically associated with stupor or nausea, in other cases - complex partial seizures with changes in consciousness and freezing; Secondary generalized attacks occur less frequently. In some patients, MRI shows no signs of hippocampal sclerosis or other abnormalities of cerebral structures. There are no pathological changes in EEG data in approximately half of the patients. Less than half of cases of familial mesial temporal epilepsy require treatment with antiepileptic drugs [3, 5, 6].
Partial (focal) autosomal dominant epilepsy with auditory stimuli
This form of the disease is actually one of the subtypes of lateral temporal lobe epilepsy; it is also known as telephone epilepsy. The disease debuts at the age of 8–19 years (most often in the second decade of life). Partial autosomal dominant epilepsy with auditory stimuli is characterized by auditory disturbances (the patient feels undifferentiated sounds and noises), auditory hallucinations (changes in the perception of volume and/or pitch of sounds, voices “from the past,” unusual singing, etc.). In addition to auditory disorders and hallucinations, this form of the disease is characterized by various autonomic disorders, pathological motor activity, as well as numerous sensory and mental disorders of varying severity. During the interictal period, EEG may show paroxysmal activity in the temporal or occipital leads (or be completely absent) [3, 5, 9].
Epilepsy with grand mal attacks on awakening
In children, the onset of seizures with this idiopathic generalized epilepsy mainly occurs in the second decade of life. In its manifestations, the disease is somewhat reminiscent of juvenile myoclonus epilepsy of Janz. The development of tonic-clonic seizures occurs exclusively or predominantly after awakening (> 90% of cases) or in the evening during the period of relaxation. Seizures are caused by sleep deprivation. In contrast to Janz myoclonus epilepsy, myoclonus and absence seizures are rarely observed in patients with this form of epilepsy. EEG makes it possible to register generalized peak-wave activity and signs of photosensitivity (the latter are not always present) [3–6, 9].
Unferricht–Lundborg disease (Baltic or Finnish myoclonus epilepsy)
This rare form of epilepsy begins in children between the ages of 6 and 13 years (usually around 10 years of age). Its manifestations resemble Ramsay Hunt syndrome. The first symptoms are seizures. Myoclonus occurs after 1–5 years; they are noted mainly in the proximal muscles of the limbs, are bilaterally symmetrical, but asynchronous. Myoclonus is induced by photosensitivity. The severity of myoclonus gradually increases. Subsequently, a progressive decline in intelligence occurs (to the point of dementia). In the later stages of the disease, patients develop signs of cerebellar ataxia [3, 5, 6].
Juvenile neuronal ceroid lipofuscinosis type III
This disease is also known as “progressive epilepsy with mental retardation” or “northern epilepsy”. It is a representative of neurodegenerative storage diseases. Debuts in preschool (5–6 years) or school (7–10 years) age. It is characterized by manifestation in the form of generalized seizures (tonic-clonic seizures) or complex partial (focal) seizures. As patients reach puberty, the frequency of attacks decreases significantly. In adulthood, it is possible to achieve complete remission of attacks [3, 5, 6, 9].
Catamenial (menstrual) epilepsy
With this type of epilepsy, which is not an independent nosological form, the occurrence of attacks is associated with phases of the menstrual cycle, influenced by numerous endogenous and exogenous factors (presumably cyclic changes in the content of sex hormones in the body, disturbances in water and electrolyte balance, the influence of the full moon, fluctuations in the level of antiepileptic drugs in blood). The disease is characterized by a clear dependence on the menstrual cycle. According to some data, generalized forms of the disease, juvenile myoclonus epilepsy and juvenile absence epilepsy occur with almost equal frequency among teenage girls. Generalized convulsive paroxysms usually tend to increase in frequency in all patients with catamenial epilepsy [3, 5, 9].
Epilepsy with an undifferentiated age range of onset
In some epilepsies, the age-related features of onset are considered unclear. The main ones are discussed below.
Epilepsy with myoclonic absence seizures
The disease most often occurs at the age of 5–10 years and is characterized by clinical manifestations in the form of absence seizures, combined with intense rhythmic bilateral clonic or (less often) tonic convulsive twitching of the proximal muscles of the upper and lower extremities, as well as the head. It is usually combined with mental development disorders and is characterized by pharmacoresistance, which determines the unfavorable prognosis of the disease. The diagnosis of epilepsy with myoclonic absence seizures is considered to be made in patients who meet the criteria for childhood absence epilepsy, but the absence seizures must be accompanied by myoclonic jerks. Treatable with valproate, ethosuximide or lamotrigine (the combination of valproate with lamotrigine or ethosuximide is considered more effective); Cancellation of treatment is possible no earlier than 2 years after achieving complete remission (clinical and instrumental) [3–5, 9].
Generalized epilepsy with febrile seizures plus
Refers to genetically determined epileptic syndromes and represents several types of epilepsy (GEFS+ type 1, GEFS+ type 2, GEFS+ type 3, GEFS+ type 5, FS with afebrile seizures and GEFS+ type 7). It is believed that in GEFS+, first described in 1997, a diagnosis of epilepsy is not necessary. Generalized epilepsy with febrile seizures plus usually occurs in children aged 1–6 years. The average age of children at the time of GEFS+ debut is about 12 months. The disease manifests itself in the form of febrile convulsions against a background of fever and in the form of other epileptic paroxysms. In addition to recurrent febrile seizures (classic tonic-clonic), GEFS+ is characterized by the presence of afebrile seizures; The clinical picture of the disease may include absence seizures, myoclonus, myoclonic-astatic and atonic seizures. In most cases, GEFS+ phenotypes turn out to be benign (once adolescence is reached, attacks are often eliminated). Upon reaching adulthood, some patients may experience rare attacks (under the influence of stress and sleep deprivation). Children with GEFS+ do not generally require antiepileptic pharmacotherapy, although some authors recommend the use of benzodiazepines (acutely or preventively). Valproate is indicated only in cases where GEFS+ persists after 6 years of age, and lamotrigine is used in cases resistant to valproate [3, 5, 6, 9].
Benign psychomotor epilepsy of childhood, or benign partial epilepsy with affective symptoms
Localization-dependent focal form of epilepsy. The etiology can be cryptogenic, familial or symptomatic. It occurs in children of all ages (7–17 years). Benign psychomotor epilepsy is characterized by recurrent seizures originating from lesions in the temporal lobe, most often from the mesial region. The disease is characterized by a wide range of mental phenomena, including illusions, hallucinations, dyscognitive states and affective disturbances. Most complex partial (focal) seizures originate in the temporal lobes. This form of epilepsy is characterized by monoform seizures, onset in childhood, and age-dependent disappearance of all clinical and EEG manifestations [3–5, 9].
Atypical benign partial epilepsy, or pseudo-Lennox syndrome
Mostly observed in children aged 2–6 years (74% of observations). At the time of the onset of the disease, approximately a quarter of children show signs of delayed speech development. In boys it debuts earlier than in girls. Characterized by generalized minor seizures (atonic-astatic, myoclonic, atpic absence seizures). A distinctive feature of the disease is the extremely pronounced activation of epileptic seizures during sleep. The main type of seizures is minor generalized (67%), with 28% of patients having simple partial seizures of the orofacial region (or generalized tonic-clonic seizures originating from the orofacial region). In addition to this, the following types of seizures occur with varying frequency in children (in descending order): generalized tonic-clonic (44%), partial motor (44%), unilateral (21%), versive (12%), focal atonic ( 9%), complex partials (2%). A small proportion of patients experience the phenomenon of epileptic negative myoclonus. The EEG pattern resembles that of rolandic epilepsy (focal sharp slow waves and peaks), but is characterized by generalization during sleep. With regard to seizures, the prognosis of the disease is favorable (all patients are “seizure-free” by the age of 15), but children often experience intellectual deficits of varying severity (about 56% of observations) [3, 5].
Aicardi syndrome
A type of epileptic syndrome associated with malformations of the central nervous system (cortical organization disorders, schizencephaly, polymicrogyria. The syndrome described by J. Aicardi et al. (1965) includes agenesis of the corpus callosum with chorioretinal disorders and is characterized by infantile flexor spasms. Affects almost exclusively girls, although known 2 cases of registration of the disease in boys with an abnormal genotype (both children had two X chromosomes).In addition to epileptic manifestations, the following pathological changes are typical for Aicardi syndrome: chorioretinal lacunar defects, complete or partial agenesis of the corpus callosum, malformations of the thoracic spine, microphthalmia , coloboma of the optic nerve, etc. Clinically, Aicardi syndrome is characterized by infantile spasms (often with an early onset) and partial (focal) epileptic seizures (in the first days or weeks of life), as well as severe retardation in intellectual development. Epileptic seizures with Aicardi syndrome are almost always turn out to be pharmacoresistant. The prognosis of the disease is unfavorable [3, 5–7, 9].
Electrical status epilepticus slow-wave sleep (ESES)
This type of epilepsy is also known by another name (continuous peak-wave discharge epilepsy during slow-wave sleep, CSWS). It is considered idiopathic epilepsy and debuts in children from about 2 years of age. Clinically characterized by focal, generalized tonic-clonic and/or myoclonic seizures that occur while awake or asleep (not always observed). Subsequently, the disease leads to speech development disorders, behavioral disorders, and cognitive dysfunctions of varying severity. The diagnosis is established on the basis of EEG data during sleep (a specific pattern in the form of continuous generalized peak-wave activity); in this case, epileptiform activity should occupy 85–100% of the total duration of the slow-wave sleep phase. At the moment of awakening, the EEG allows you to register the presence of sharp waves. The prognosis in terms of the disappearance of epileptic seizures and EEG changes is relatively favorable (by puberty), but cognitive impairment remains in children [3–6].
Frontal nocturnal autosomal dominant epilepsy
Refers to isolated epileptic syndromes. Debuts before the age of 20 (usually around 11 years of age). Seizures occur when falling asleep and/or waking up (in the form of short - up to 1 minute, episodes of hyperkinesis, with or without loss of consciousness); epileptic seizures are preceded by an aura (a feeling of fear, trembling or somatosensory phenomena). Secondary generalized seizures occur in 50–60% of patients; In about a quarter of cases, attacks occur while awake.
Ictal EEG studies record sharp and slow waves or rhythmic low-voltage fast activity in the frontal leads. In the interictal period, EEG data may be normal or periodically show peaks in the frontal leads [3, 5, 9].
Conclusion
Among the epilepsies that occur in children of any age (0–18 years), it is necessary to list Kozhevnikovsky epilepsy (chronic progressive partial epilepsy, or epilepsia partialis continua), the clinical manifestations of which are well known to pediatric neurologists, as well as localization-related forms of epilepsy (frontal, temporal , parietal, occipital) [3]. The latter belong to symptomatic and probably symptomatic focal epilepsies, in which a wide range of symptoms is determined by the localization of the epileptogenic focus.
Read the continuation of the article in the next issue.
Literature
- Brown T. R., Holmes G. L. Epilepsy. Clinical guidelines. Per. from English M.: Publishing house BINOM. 2006. 288 p.
- Mukhin K. Yu., Petrukhin A. S. Idiopathic forms of epilepsy: systematics, diagnosis, therapy. M.: Art-Business Center, 2000. 319 p.
- Epilepsy in neuropediatrics (collective monograph) / Ed. Studenikina V. M. M.: Dynasty, 2011, 440 p.
- Child neurology (Menkes JH, Sarnat HB, Maria BL, eds.). 7 th ed. Lippincott Williams & Wilkins. Philadelphia-Baltimore. 2006. 1286 p.
- Epileptic syndromes in infancy, childhood and adolescence (Roger J., Bureau M., Dravet Ch., Genton P. et al, eds.). 4 th ed. (with video). Montrouge (France). John Libbey Eurotext. 2005. 604 p.
- Encyclopedia of basic epilepsy research/Three-volume set (Schwartzkroin P., ed.). Vol. 1–3. Philadelphia. Elsevier/Academic Press. 2009. 2496 p.
- Aicardi J. Diseases of the nervous system in children. 3rd ed. London. Mac Keith Press/Distributed by Wiley-Blackwell. 2009. 966 p.
- Chapman K., Rho JM Pediatric epilepsy case studies. From infancy and childhood through infancy. CRC Press / Taylor&Francis Group. Boca Raton–London. 2009. 294 p.
- Epilepsy: A comprehensive textbook (Engel J., Pedley T.A., eds.). 2nd ed. Vol. 1–3. Lippincott Williams&Wilkins/A Wolters Kluwer Business. 2008. 2986 p.
V. M. Studenikin, Doctor of Medical Sciences, Professor, Academician of the Russian Academy of Economics
FSBI "NTsZD" RAMS, Moscow
Contact Information
Manifestations of epileptic seizures
Epilepsy is characterized by uncontrolled spontaneous seizures, the occurrence and intensity of which cannot be predicted in advance. The disease can be suspected after the first attack occurs. If the attack recurs, you should immediately consult a neurologist, since in the absence of proper treatment, the frequency of epileptic seizures only increases over time.
The aura can be manifested by the following sensations:
- certain sounds (ringing or tinnitus, melody, other sounds);
- some odors;
- visual visions;
- a feeling of deja vu;
- headaches;
- increased anxiety, etc.
In reflex epilepsy, the appearance of seizures is provoked by a certain irritant. An epileptic seizure in such cases can begin as a result of exposure to bright light, a flash of light, light music in a nightclub, or a fast video sequence with a frequently changing picture.
In addition, epilepsy may be characterized by the following symptoms:
- convulsions: during an attack, tonic or clonic convulsions appear in the muscles of the upper and lower extremities;
- disorder of consciousness: some simple epileptic seizures are accompanied by the preservation of consciousness, others by turning it off or the patient falling into a coma. In addition, during an epileptic seizure, an epileptic may see hallucinations and mentally move to another place;
- autonomic disorders: during an attack, patients experience changes in pulse, blood pressure, and vascular tone;
- memory impairment: after the end of an epileptic attack, the patient most often cannot remember what happened to him. As the disease progresses and the frequency of attacks increases, epileptics experience impaired speech and thinking, which leads to dementia.
In the early stages of the disease, as a rule, simple epileptic seizures occur, characterized by a short duration and the absence of loss of consciousness. As the disease progresses, the condition worsens and attacks become more frequent with more noticeable consequences.
What are the types of seizures?
Along with classic grand mal seizures, there are also so-called petit mal seizures, which manifest themselves in short-term, up to several seconds, loss of consciousness.
The patient does not fall to the floor. Convulsions are mild. The attack is accompanied by a violent reaction from the internal organs and skin. A cataleptic seizure often occurs during conditions associated with great emotional stress, sometimes even during laughter. The child falls, but not sharply, but due to decreased muscle tone, he seems to sag and become limp. During a seizure, the patient’s consciousness is completely preserved; memory of everything that happens during this event is not lost.
Narcoleptic seizure . Suddenly, an extremely strong, irresistible state of drowsiness occurs. The sleep that follows is usually short, but deep; patients often fall asleep in the most unusual positions and in the most unexpected places. After awakening, the normal state is completely restored. All mental processes return completely to normal. The child feels well rested, cheerful and full of energy.
A hysterical attack occurs, firstly, always against the background of some kind of mental trauma, and secondly, always in the presence of strangers. Consciousness during an attack may be impaired, but not severely, and is never completely absent. The patient falls to the floor, but not abruptly; the fall is always careful, while the child tries not to collide with sharp and hard objects. During such a descent, he gives the impression of being exhausted.
The seizure lasts much longer than all other types - 30 minutes or more. Most often, the student rolls on the floor or on the bed, knocks his hands and feet on the floor, bends in the form of an arc, begins to shake all over, screams loudly, moans, and cries. Unlike a grand mal seizure, involuntary urination and defecation are never observed.
Types of disease
The following types of epilepsy are observed in patients of different age groups.
- Juvenile myoclonic epilepsy. Mostly occurs in teenagers. The attacks occur after waking up and are sometimes not accompanied by convulsions.
- Progressive myoclonic epilepsy. It is difficult to treat and can develop into a more complex and dangerous form of the disease throughout life.
- Symptomatic epilepsy. Older people suffer. The first signs of the disease may appear up to 30 years of age. The causes are head injuries and diseases of the nervous system. Due to the varied manifestations of the disease, it is difficult to diagnose as the symptoms can be misleading and the patient may be misdiagnosed even after a thorough examination.
- Temporal lobe epilepsy. Affects the temporal lobes of the brain. A common symptom is a state of “déjà vu.” This form of epilepsy also causes anxiety disorders, uncontrollable outbursts of anger and other emotional states.
- Frontal epilepsy. Symptoms vary and depend on which parts of the brain are affected. Affects the motor functions of the human body. The manifestations of this disease often do not cause concern. The patient may move his eyes and tongue rapidly, and stagnate. From the outside it may seem that a person is simply experiencing nervous excitement. At the same time, a person cannot organize his thoughts, experiences many emotions at the same time and cannot calm down or concentrate on one thing.
- Parietal epilepsy. It is quite rare. The symptom is visual disturbances or eye sensitivity to flashes of light. The eyes may move uncontrollably or jerk from side to side, and the eyelids may tremble. Severe headaches often occur during or after an attack.
- Absence epilepsy. Symptoms of this disease are characterized by short-term fainting or loss of consciousness. With rare exceptions, absence epilepsy is manifested by a loss of concentration; a person cannot focus, breaks off a sentence mid-sentence and cannot build a logical chain to convey his idea. His movements are unclear, his hands can move uncontrollably, inexplicable sensations arise in his fingers, so the patient wants to stretch his hands.
- Myoclonic epilepsy. Manifests itself with sudden spontaneous movements of the arms and legs. The symptoms of this disease are often ignored and confused with the completely normal hypnogogic myoclonus that everyone experiences when falling asleep.
In addition to the classification of epilepsy, individual forms of epileptic seizures are also distinguished.
Types and forms of epilepsy in children
Doctors around the world are studying epilepsy closely. There are extremely rare forms of manifestations and symptoms, but most often the disease is characterized by certain types and forms:
- Rolandic epilepsy. The name of the disease is due to the fact that the focus of the disease is located in the central (Rolandic) sulcus of the brain. During a seizure, the child feels tingling and numbness in the facial muscles. Speech disappears for a while and active salivation begins. At the same time, the child remains conscious. The seizure usually lasts no more than 5 minutes. Most often, children aged 3 to 13 years are affected, and the disease recedes from the onset of puberty.
- Nocturnal epilepsy. This form of the disease affects only children under 3 years of age. The disease manifests itself exclusively at night. The attack passes without pain syndromes. Epilepsy is expressed in enuresis (urinary incontinence), sleepwalking, limb cramps during sleep, and irritability develops.
- Absence form of epilepsy. The mildest type of the disease is observed in children aged 5 to 8 years. As a rule, after 8 years the disease completely disappears, rarely becoming more severe. The main symptoms of absence epilepsy: memory lapses during an attack, gaze frozen at one point, synchronous uncontrolled rotation of the head and limbs.
- Idiopathic form of epilepsy. The child has no deviations in either physical or intellectual development. With this type of disease, the patient suffers from periodic seizures with loss of consciousness, increased salivation, and interruptions in breathing. After a seizure, the child has no memory of what happened.
Diagnosis of the disease at the Yusupov Hospital
No doctor can determine a reliable diagnosis after one single attack, since an epileptic seizure can occur once and in completely healthy people.
The following modern methods are used to carry out diagnostics at the Yusupov Hospital:
- computed and magnetic resonance imaging;
- angiography;
- electroencephalography;
- neuroradiological diagnostics;
- examination by an ophthalmologist of the fundus;
- biochemical blood test.
In some cases, a lumbar puncture is prescribed - a test that allows you to identify an infection that has affected the brain.
Diagnosis and treatment of epilepsy
The purpose of diagnosis for suspected epilepsy is to examine the bioelectrical functioning of the brain. To do this, the patient is prescribed the following diagnostic measures:
- daily EEG monitoring;
- MR angiography to exclude cerebral aneurysm;
- magnetic resonance imaging (MRI) for suspected injuries, hematomas, tumors or abscesses;
- EEG upon awakening.
Treatment of epilepsy in adolescents is complex. The duration is on average 3-5 years. In some cases, lifelong medication and a special keto diet are prescribed. Therapy involves following a special regime: avoiding lack of sleep, excessive stress, and bad habits. Medicines are needed to keep myoclonic seizures under control. If they are completely suppressed, the person will not be able to understand that he is about to have a seizure.
Recently, patients are increasingly being prescribed new drugs, the effectiveness of which has already been tested in clinical settings. Only one of the anticonvulsants is used. It is selected by monitoring the body's reaction.
A combination of 2-3 drugs is practiced only for severe forms of epilepsy. In such cases, side effects may occur. Reception begins with minimal doses, gradually increasing them until the optimal dosage is found.
The question of whether it is possible to completely cure epilepsy in adolescents cannot be answered with certainty. Each specific case will have its own forecast. It all depends on the cause that caused the disease, the state of the nervous system and brain, as well as the treatment regimen. But in many cases, after 1-3 years of therapy, the diagnosis is removed.
First aid in case of attack:
- lay the patient on the floor to avoid falling and hitting his head;
- turn on its side so that the child does not choke on vomit or saliva;
- hold your head to avoid injury;
- do not leave until the child fully regains consciousness.
Under no circumstances should you try to open your mouth and hold your tongue. With your mouth open, you can put a bandage between your teeth, but so that nothing interferes with normal breathing.
Treatment at the Yusupov Hospital
To alleviate the condition of patients suffering from epilepsy, the specialists at the neurology clinic at the Yusupov Hospital have several methods at their disposal. Properly selected treatment and strict adherence by the epileptic to medical recommendations can achieve stable remission for a long time.
Drug treatment helps to reduce electrical activity in the lobe of the brain where the disease, previously identified through EEG (electroencephalography), is localized. Treatment of patients at the Yusupov Hospital is carried out using the most modern medications that have a pronounced therapeutic effect and have minimal side effects. During drug treatment at the Yusupov Hospital, patients are constantly monitored by doctors who monitor the course of the disease.
Methods of treatment or correction
Methods of treatment and correction of the disease are individual in nature. However, it is mandatory to obtain the results of the following studies:
- CT and MRI of the brain;
- encephalography (EEG);
- daily EEG monitoring;
- EEG of night sleep.
The data obtained is analyzed by a neurologist and, based on them, a plan for treatment or correction of the disease is developed. Complex treatment may be required for 2-4 years. In some cases, the use of medications becomes lifelong.
In addition to taking medications, your doctor may recommend:
- follow a diet that excludes foods contraindicated for this disease;
- strict daily routine;
- visiting a psychologist.
The main task of medications is to reduce the regularity of attacks and stop the progression of the disease. It is possible to stop the development of the disease in children in 70% of cases. A complete recovery from epilepsy is achieved in 30% of cases.
In what cases is hospitalization required?
The patient must be taken to the hospital:
- if epilepsy was detected for the first time - to conduct research and select effective treatment;
- with status epilepticus;
- in case of planning surgical intervention (in particular, removal of a brain tumor accompanied by epileptic seizures);
- in order to routinely assess the dynamics of the disease.
Physical therapy exercises, supervised by an experienced physical therapy doctor, can normalize the processes of excitation and inhibition in the brain, which are most often disturbed in epileptics. Special rhythmic movements and breathing exercises have a positive effect on neurons, harmonize the mental state of patients, and prevent stress and other diseases.
Prevention of epileptic seizures
There are preventive recommendations that, if followed, can prevent epileptic seizures, especially if you already have a history of them:
- you should protect yourself from getting traumatic brain injuries;
- stop smoking, drinking alcohol and drugs;
- try not to stay in stuffy, unventilated rooms for a long time;
- avoid hypothermia;
- give preference to a healthy lifestyle and sports;
- avoid stressful situations;
- do not overwork;
- observe the work and rest schedule.
Epilepsy is a rather severe and difficult-to-treat pathology. Thanks to the prescription of modern anticonvulsants, doctors at the neurology clinic of the Yusupov Hospital are able to significantly improve the quality of life of their patients and prevent the development of new epileptic seizures in them.
An appointment with a neurologist at the Yusupov Hospital can be made by phone or on the clinic’s website. You can also ask questions regarding the conditions of hospitalization in a hospital and the estimated cost of the services provided.