Charcot-Marie-Tooth disease belongs to a group of heterogeneous genetic diseases that are characterized by damage to the peripheral nervous system with the development of atrophy of the muscles of the limbs. Along with this, there is a loss of sensitivity, decreased tendon reflexes and deformation of the limbs. The disease is genetically heterogeneous; about 40 mutations localized in more than 20 genes can cause it, but the most common mutations are in the PMP22, MPZ, GJB1, MFN2 genes. Inheritance most often occurs according to an autosomal dominant type, but other options are possible, including linkage to the X chromosome.
Pathogenetic aspects of Charcot-Marie-Tooth disease and its symptoms
The pathogenesis of Charcot-Marie-Tooth disease is based on nerve damage, which leads to secondary muscle atrophy. Most often, the “fast” motor nerve fibers that innervate the remote muscles of the extremities that take on greater physical activity—the muscles of the foot and lower leg—are affected. The muscles of the hands and forearms are damaged a little less and later. Sensory nerves are also affected, which leads to loss of tactile, pain and temperature sensitivity of the limbs.
Typically, Charcot-Marie-Tooth disease manifests itself between the ages of 10 and 20 years. First, symmetrical weakness in the legs occurs, which leads to a characteristic change in gait like a steppage, or, as it is also called, “rooster gait.” Then the feet begin to turn in and become deformed, their arch increases and a hollow foot is formed. As muscle atrophy progresses, the legs take on the appearance of inverted bottles.
Gradually, damage to the hands occurs: due to atrophy of the muscles of the hand, it becomes like a monkey’s paw. In addition, sensitivity suffers, primarily superficial. Sometimes there is cyanosis of the affected limbs and swelling in them.
Charcot-Marie-Tooth disease has a slow course. Sometimes the period between manifestation and hand atrophy lasts 10 years. Even despite the development of atrophy, patients retain the ability to work and self-care for quite a long time. With proper care, life expectancy does not suffer and remains at the general population level.
Publications in the media
Charcot–Marie–Tooth disease combines hereditary sensorimotor neuropathies types I and II. A disease characterized by weakness and atrophy of the distal group of muscles of the lower extremities and usually an autosomal dominant type of inheritance; often combined with other neurodegenerative diseases (for example, Friedreich's ataxia).
Genetic aspects and classification. Types and genes: • type 1A: 118220, PMP22, CMT1A, 601097 (myelin protein 22), 17p11.2; • type 1B: 118200, MPZ, CMT1B, 159440, 1q22; • type 2A: CMT2A, 118210, 1p36 p35; • type 2B: CMT2B, 600882, 3q13 q22; • type 2D: CMT2D, 601472, 7p14; • type 4A: CMT4A, 214400, 8q13 q21.1; • type 4B: CMT4B, 601382, 11q23; • X linked 1, dominant: 302800, GJB1, CMTX1, 304040 (connexin gene CX32, Xq13.1); • X linked 2, recessive: CMTX2, 302801, Xp22.2; • demyelinating: CMTND, 601596, 5q23 q33; • with skin aplasia (302803) • with palmoplantar keratoderma and nail dystrophy (148360) • Couchok's syndrome (310490) • with Friedreich's ataxia (302900) • with deafness (118300, 214370) • with tremor (214380) • with ptosis and parkinsonism (118301)
Pathomorphology • Hereditary sensorimotor neuropathy type I - histologically segmental demyelination and remyelination, concentric growth of Schwann cells (hypertrophic neuropathy) • Hereditary sensorimotor neuropathy type II - histologically Waller's degeneration.
Clinical picture • Hereditary sensorimotor neuropathy type I •• Onset in middle childhood •• Weakness of the foot extensors (foot drop) •• Slowly progressive atrophy of the distal muscle groups of the legs (stork legs) •• Atrophy of the hand muscles develops later •• Decreased vibration, pain and stocking-type temperature sensitivity •• Tendon reflexes decrease and disappear •• Thickened nerves are sometimes palpable •• Abnormally enlarged arch of the foot (pes cavus) is often the only symptom in heterozygous carriers of the defective gene •• The course is slowly progressive, the disease has virtually no effect on lifespan • Hereditary sensorimotor neuropathy type II •• Onset of muscle weakness between 16 and 30 years of age •• The disease progresses more slowly than Charcot-Marie-Tooth disease type I.
Laboratory studies • Charcot-Marie-Tooth disease type I •• Decreased nerve conduction velocity •• Prolongation of distal latency • Charcot-Marie-Tooth disease type II •• Nerve conduction velocity is usually normal •• Decreased amplitude of evoked potentials.
Treatment • There is no specific treatment • The choice of profession should be made taking into account the slow progression of the disease • Braces are used for foot drop • Orthopedic correction of the foot.
Synonyms •• Charcot-Marie muscular atrophy •• Muscular atrophy of the peroneal type •• Hereditary neural amyotrophy
ICD-10 • G60.0 Hereditary motor and sensory neuropathy
Classification of Charcot-Marie-Tooth disease
The genetic classification is very extensive, since the development of the disease is based on about 40 mutations affecting more than 20 different genes. Based on morphological characteristics and electromyographic data, three main types of Charcot-Marie-Tooth disease are distinguished:
- demyelinating type. It is characterized by destruction of the myelin sheath and, as a consequence, a decrease in impulse conduction velocity (ISV) along the median nerve;
- axonal type. Normal or slightly reduced SPI along the median nerve is typical, since axons are primarily affected;
- intermediate type. The speed of impulse conduction has borderline values.
This classification makes sense for narrowing the search range in genetic diagnosis, since certain mutations are characterized by their own clinical manifestations.
International classification of neural amyotrophy
A number of pathologies with motor and sensory lesions belong to the group of hereditary amyotrophies. The international classification ICD 10 codes the nosology - “G12”. Hereditary transmission makes etiological treatment impossible. A complex of common disorders including deafness, retinitis, and ataxia is treated with supportive medications.
List of neural muscular amyotrophies:
- Charcot-Marie-Tooth;
- Dezherina-Sotta – hypertrophic changes in the sheath of nerve fibers cause a gradual increase in clinical symptoms caused by sensorimotor neuropathy;
- Degenerative changes in the trunk and horns of the spinal cord occur in three clinical forms - late, early, congenital. The nosology is called “Werdnig-Hoffmann spinal muscular atrophy”;
- The destruction of motor neurons with subsequent atrophic disorders of the spinal horns is characterized by muscle twitching. The pathology is called “Kullberg-Welander atrophy”;
- Other amyotrophies are hereditary - neurogenic malignant, scapulofibular, New England.
According to the international classification of diseases, symptoms with similar clinical signs should be distinguished. Related manifestations are treated according to other regimens, so differential diagnosis is required.
Variants of Charcot-Marie-Tooth syndrome:
- The first type is accompanied by a change in the characteristics of nerve signal transmission against the background of destruction of the myelin sheaths;
- The second option is that the impulse remains, but damage to the axons of Schwann cells is detected.
According to Wikipedia, it is necessary to verify the morphological forms of the disease at an early stage before complications develop.
Treatment of Charcot-Marie-Tooth disease
To date, no cure for Charcot-Marie-Tooth disease has been developed. Symptomatic therapy is used to improve muscle trophism. For this purpose, vitamins, ATP, glucose, cocarboxylase are prescribed, and physiotherapeutic treatment, exercise therapy, and massages can also be used. In some cases, orthopedic and spa treatment is necessary.
In medical genetics, a search is carried out for most of the mutations leading to the development of Charcot-Marie-Tooth disease. We use DNA sequence sequencing as a research method.
What is Charcot-Marie-Tooth neural amyotrophy?
The chronic course of the disease is accompanied by gradual progression of muscle paresis and paralysis. The pathology is accompanied by changes in muscles with gradual atrophic changes in the legs at the initial stage, secondary atrophy of the arms. Fascicular twitching occurs due to a disruption in the transmission of cholinesterase in nerve fibers due to an infectious lesion. Maintenance treatment is aimed at restoring antioxidant and metabolic disorders.
A decrease in nutrient intake is characterized by atrophic changes in muscles and loss of contractile function. Pathogenetic mechanisms are caused by the destruction of motor neurons of the spinal cord, stem structures, and anterior intracerebral horns.
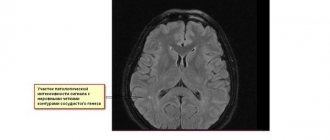
MRI of multifocal leukoencephalopathy
Risk factors and causes of CMT
CMT is a hereditary disease, so people who have close relatives with the disease have a higher risk of developing the disease.
The disease affects peripheral nerves. Peripheral nerves are made up of two main parts: the axon, which is the inner part of the nerve, and the myelin sheath, which is the protective layer around the axon. CMT can affect the axon and myelin sheath.
At CMT 1
genes that cause the breakdown of the myelin sheath are mutated. Eventually, the axon is damaged and the patient's muscles no longer receive clear messages from the brain. This leads to muscle weakness and loss of sensation or numbness.
For CMT 2
the mutating gene directly affects axons. The signals are not transmitted strongly enough to activate the muscles and senses, so patients have weak muscles, poor sensation, or numbness.
ShMT 3
or
Dejerine-Sottas disease
, a rare type of disease. Damage to the myelin sheath leads to severe muscle weakness and soreness. Symptoms may be noticeable in children.
ShMT 4
is a rare disease that affects the myelin sheath. Symptoms usually begin in childhood and patients often require a wheelchair.
ShMT X
caused by a mutation on the X chromosome. It is more common in men. A woman with CMT X will have very mild symptoms.
How to diagnose CMT?
The doctor will ask about your family history and look for signs of muscle weakness - decreased muscle tone, flat feet, or high arches (cavus).
Nerve conduction studies measure the strength and speed of electrical signals that travel through nerves (electromyography). Electrodes are placed on the skin and deliver mild electric shocks that stimulate the nerves. A delayed or weak response suggests a neurological disorder, and possibly CMT.
With electromyography (EMG), a thin needle is inserted into the muscle. When the patient relaxes or contracts the muscles, electrical activity is measured. Testing different muscles will show which one is suffering.
Genetic testing is done using a blood sample that can show whether a patient has gene mutations.
Symptoms of CMT in adults
- Weakness in the muscles of the legs and ankles;
- Curvature of toes;
- Difficulty lifting the foot due to weak ankle muscles;
- Numbness in the arms and legs;
- Change in the shape of the lower leg, with the leg becoming very thin below the knee, while the thighs retain normal muscle volume and shape (stork leg);
- Over time, the arms become weaker and patients find it difficult to perform daily tasks;
- Pain in the muscles and joints appears, and it is difficult for a person to walk. Neuropathic pain occurs due to damaged nerves;
- In severe cases, the patient may need a wheelchair, while others may use special shoes or other orthopedic devices.
Clinical symptoms of Charcot-Marie-Tooth amyotrophy
Initially, weakness is localized in the lower extremities. Muscle twitching makes it impossible for a person to stay in one place. To reduce pain during paresthesia, the patient begins to mark time. Tingling in both lower extremities is not relieved by medications. Bilateral muscle atrophy causes a specific appearance of the foot - “stork limb”, “inverted bottle symptom”. A complex of changes causes deformation of the lower limb. A person will not be able to stand on his heel. A high arch causes the leg to be constantly raised. Walking is difficult.
Atrophic changes in the upper extremities are consistently observed. Difficulties in transmitting nerve impulses to the small muscles of the arm, symmetrical atrophy of the hypotenor region complement the diagnostic range characteristic of the nosological form. Increased muscle tone causes a specific appearance of the upper limbs - “monkey”, “clawed”.
Neurologists record the pathology of tendon reflexes:
- Achilles;
- Knees;
- Biceps brachii muscle.
Amyotrophic disorders are accompanied by sensory changes. First, the surface receptors are upset, and the “socks and gloves” syndrome is formed, in which the sensitivity of the feet and hands is lost. At the same time, skin hyperemia and hyperhidrosis of the extremities occur. Excessive sweating is a secondary symptom caused by loss of innervation of the sweat glands. The patient's intellectual function is preserved.
Amyotrophic Charcot-Marie-Tooth dysfunction is accompanied by disorders of the extensors and abductors of the feet. The gait resembles that of a horse due to Friedreich's high foot. X-ray of the lower extremities reveals hammertoe deformity.
The progression of the disease is long. About ten years pass from the appearance of the first signs to atrophy of the arm muscles. Amyotrophic changes are accompanied by exogenous disorders.
Provoking factors of the disease:
- Hypothermia;
- Acute respiratory viral infection;
- Rubella;
- Infectious mononucleosis;
- Measles;
- Hypovitaminosis.
An autosomal recessive or autosomal dominant type of genealogical analysis linked to the X chromosome makes it possible to distinguish the nosology from myopathic Govers-Welander syndrome, bacterial polyneuritis, and myotonic dystrophy.