История заболевания и номенклатура
Синдром доброкачественной миоклонической эпилепсии у детей (СДМЭД) не имел четкого определения до первого описания его у 7 детей младшего возраста в 1981 г. (Драве и Биор, 1981). В этой статье СДМЭД определялся как случай миоклонических припадков (МП) без припадков другого типа, кроме простых фебрильных припадков (ФП), в первые три года жизни у здоровых детей. Эти МП легко поддавались лечению и становились достаточно редкими в течение дальнейших лет периода детского возраста. Психомоторное развитие оставалось нормальным, каких-либо тяжелых психологических последствий не наблюдалось. С тех пор в литературе было описано много других случаев. СДМЭД включен в Международную классификацию 1989 г. среди генерализованных идиопатических эпилепсий (Комиссия, 1989). Некоторые авторы описали у больных рефлекторный МП, вызываемый шумом или разговором, и предложили различать две отдельные нозологические формы, причем 2-я была названа рефлекторной миоклонической эпилепсией у детей (Риччи и др., 1985). Мы думаем, что такое деление нецелесообразно и будем описывать все случаи как СДМЭД.
Насколько нам известно, в литературе на сегодняшний день описано 98 случаев, из которых 88 соответствуют классическому описанию СДМЭД, а 10 определялись как «рефлекторная СДМЭД».
Сейчас под нашим наблюдением находятся 5 новых больных, четверо из которых со спонтанными и рефлекторными припадками, что доводит общее число больных до 103.
В первом описании СДМЭД указывалось, что болезнь начиналась до 3-летнего возраста, тогда как в последующих сообщениях говорилось о более позднем начале у некоторых больных — до 4 лет 8 месяцев (Джиованарди-Росси и др., 1997). Это означает, что эпилепсия одного типа может появиться в разном возрасте (Гуэрини и др., 1997).
Общие сведения
Юношеская миоклоническая эпилепсия (ЮМЭ) составляет до 12% от всех форм этого заболевания и около 23% случаев идиопатической генерализованной эпилепсии. ЮМЭ является одной из разновидностей миоклонической эпилепсии — генерализованной эпилепсии, протекающей с миоклоническими приступами. В эту группу заболеваний также входят: детская доброкачественная миоклония, синдром Веста (миоклоническая энцефалопатия детского возраста с церебральной гипсаритмией), болезнь Лафоры и др.
Первое описание ЮМЭ датировано 1867 г. Однако в качестве отдельной нозологической единицы юношеская миоклоническая эпилепсия была выделена лишь в 1955 г. по предложению группы германских врачей во главе с Янцем, после чего она стала упоминаться как синдром Янца. В научной литературе по неврологии и эпилептологии можно встретить также термин «импульсивный petit mal».
Юношеская миоклоническая эпилепсия
Общие положения
Эпидемиология
Согласно немногочисленным имеющимся в наличии эпидемиологическим данным, СДМЭД составляет менее 1% всех эпилепсий (Центр Св. Павла, 1997), 1,3% и 1,72% эпилепсий, которые начинаются в первый год жизни (Далла Бернардина и др., 1983), и 0,39% эпилепсий, начинающихся в первые 6 лет жизни (Охтсука и др., 1993).
Пол
Распределение по полу: 52 мальчика и 27 девочек.
Генетика
Генетика СДМЭД неизвестна. Больных мало, а случаи семейного СДМЭД описаны не были. В семейном анамнезе эпилепсия или фебрильные припадки (ФП) присутствуют у 39% из 80 больных. У 70 больных заболеваемость ФП в семье составляла 17%, а заболеваемость эпилепсией — 24%. Трудно оценить тип эпилепсии, обнаруживаемый у родственников. В 10 случаях это, вероятно, была идиопатическая эпилепсия. В случае, описанном Арзиманоглоу и др. (1996), пробандом был 2-й из 2 братьев, а старший страдал типичной эпилепсией с миоклонически-астатическими припадками (ЭМАП, синдром Дуза).
Анамнез жизни
В анамнезе большинства больных патологии до начала МП не было. Только у двух (1,9%) были сопутствующие заболевания: синдром Дуза (Драве и др., 1992) и гиперинсулиновый диабет (Коламария и др., 1987).
Однако случаи ФП не являются редкими: 19 больных из 64 (30%). ФП были всегда простыми, однако редкими (1-2) и наблюдались до наступления миоклоний и до начала лечения (15 случаев).
Причины появления
ЮМЭ является тяжелой наследственной патологией, которая не развивается в случае органических, химических или травматических поражений. Возникновение заболевания провоцируется генетическим сбоем, точная причина которого до сих пор не установлена. Существует несколько рабочих гипотез позволяющих объяснить причины возникновения генетического сбоя:
- Согласно первой, развитие эпилепсии провоцирует дефектная хромосома в структуре гена q 15.
- Согласно второй, эпилепсию вызывает повреждение одного из локусов плеча 6 хромосомы.
- Согласно третьей (наиболее вероятной), заболевание провоцируется дефектом в строении гена C6orf33, а также BRD2.
Клинические и ЭЭГ проявления
Возраст, в котором начинается заболевание, — обычно от 4 месяцев до 3 лет. Редко начало более раннее. О более позднем начале сообщили Джиованарди-Росси и др. (1997) — 4 года 9 месяцев — и Лин и др. (1998) — 3 года 9 месяцев.
Вначале МП бывают короткими, чаще всего редкими, с вовлечением верхних конечностей и головы, редко — нижних конечностей. У младенцев они едва заметны, и родителям иногда бывает трудно определить их начало и частоту. Они часто говорят о «спазмах» или «кивании головой». Позже частота приступов возрастает.
Записи видео-ЭЭГ позволили нам провести точный анализ этих приступов. Это более или менее массивные миоклонические подергивания, поражающие туловище и конечности, провоцирующие наклон головы и движение верхних конечностей вверх и кнаружи со сгибанием нижних конечностей, а иногда — вращением глазных яблок. Их интенсивность у каждого ребенка своя, у одного и того же пациента она может меняться при каждом приступе. При тяжелых формах отмечается резкое выпадение предметов из рук, иногда больной падает. При формах средней тяжести отмечается только короткое движение головы вперед или даже простое закрытие глаз, как правило, припадки очень короткие (1-3 сек.), хотя они могут длиться и дольше (у детей старшего возраста) и состоять из псевдоритмичных подергиваний, длящихся не более 5-10 сек. Они развиваются несколько раз в день через разные промежутки времени. В отличие от детских спазмов, они не протекают длинными сериями и развиваются не в момент пробуждения, а, скорее, при дремоте. У некоторых больных вызвать припадки может резкий шум, внезапный контакт или повторная фотостимуляция (ИФС). При единичных припадках оценить состояние сознания трудно. Только при повторных припадках наблюдается легкое нарушение сознания без прерывания деятельности. Мы не наблюдали внезапных коротких вокализмов, о которых сообщает Лин и др. (1998). Авторы указывают на вовлечение в патологический процесс диафрагмы и / или абдоминальных мышц, вызывающих экспираторный шум. При рефлекторном МП миоклонус может быть вызван как в состоянии бодрствования, так и сна, порог возбудимости низкий на стадии 1, но он постепенно повышается во время медленных стадий (Риччи и др., 1995). У больных с рефлекторным МП фаза быстрого сна не записывалась и не анализировалась. Т.к. развитие ребенка идет нормально, родители и педиатры принимают эти движения за патологию.
Картина ЭЭГ вне приступа в пределах нормы.
Миоклонии всегда связаны с ЭЭГ разрядом. Записи показывают, что миоклонии сопровождаются разрядом быстрых генерализованных спайк-волн (СВ) или полиспайк-волн (ПСВ) частотой более чем 3 Гц, с той же продолжительностью, что и миоклонии. Это более или менее регулярный разряд, и он может начинаться в лобных областях и в темени. Миоклонии короткие (1-3 сек.) и обычно изолированные. За миоклоническим подергиванием может последовать короткая атония. Иногда после приступа появляются произвольные движения, расцениваемые как нормальное сокращение мышц. И только у одного больного мы отметили сочетание миоклоний в дельтовидной мышце с чистой атонией шейных мышц. Во время сонливости миоклонии усиливаются, но не всегда. Они исчезают во время медленного сна. МП, вызванные тактильными и слуховыми раздражителями, имеют одни и те же характеристики. Риччи и др. (1995) указывают на то, что первичные проявления обычно, но не всегда, проявляются только симптомом моргания, за которым через 40-80 мсек. следует миоклоническое подергивание руки. После миоклонического приступа наступает рефрактерный период, длящийся от 20-30 сек. до 1-2 мин, во время которого внешние стимулы, даже испуг, не провоцируют приступы. Повторная фотостимуляция (ПФ) тоже может спровоцировать МП (Драве и др., 1992; Тодт и Мюллер, 1992; Джиованарджи-Росси и др., 1997; Лин и др., 1998).
Данные интериктальной ЭЭГ без патологии. Спонтанные разряды СВ редки; в центральных областях мозга можно обнаружить медленные волны. ПФ вызывает СВ без имеющейся сопутствующей миоклонии. Записи короткого дневного сна показали нормальную организацию сна, генерализованные разряды СВ могут появляться во время быстрого сна.
Медицинские интернет-конференции
Ювенильная миоклоническая эпилепсия (ЮМЭ) впервые была описана как синдром Janz и Christian (1957) под названием Impulsive Petit Mal. ЮМЭ сейчас считается классическим примером идиопатической генерализованной эпилепсии. В то же время нередко допускаются ошибки в диагностике ЮМЭ, связанные с тем, что миоклонические приступы остаются незамеченными или неправильной трактовкой результатов ЭЭГ.
ЮМЭ ставляет коло 5-11% всех регистрируемых форм эпилепсии (Alfradique & Vasconcelos, 2007). Наиболее часто дебют заболевания приходится на ранний либо поздний пубертатный периоды (Janz, 1985).
ЮМЭ обычно начинается в пубертатном периоде у здоровых людей. Первый приступ часто провоцируется нарушением режима сна, употреблением алкоголя. Во многих случаях приступы развиваются вскоре после пробуждения. Также при этом нередко в семейном анамнезе выявляется наличие эпилепсии.
При ЮМЭ может развиваться три типа приступов.
1) Билатеральные миоклонические приступы. Они состоят из единичных или коротких аритмичных подергиваний рук, иногда и нижних конечностей, обычно при ясном сознании. Подергивания настолько внезапны, что пациент может ронять вещи, которые держит в руках. Пациент и его родственники часто относятся к этим приступам не как к патологии, а, скорее, как к тремору или нервозности. Если существует только такой тип приступов, то они часто остаются недиагностированными. Миоклонии могут возникать в разное время суток без четкой приуроченности к пробуждению; у некоторых пациентов встречаются только миоклонии век (Мухин К.Ю., 2000).
2) Генерализованные тонико-клонические приступы (ГТКП). Приблизительно в 1/3 случаев ГТКП являются первыми появившимися приступами, а миоклонические приступы присоединяются позже. Связь между этими приступами становится очевидна у пациентов, у которых за несколько секунд или минут до ГТКС появляются миоклонические подергивания (клонико-тонико-клонический приступ). Некоторые пациенты расценивают их как предвестники и предпринимают меры, чтобы обезопасить себя от повреждений, например не выходят из дома, ложатся или вызывают родственников. В том случае, если развитию развернутого судорожного приступа не предшествуют миоклонии, аур не бывает.
3) Абсансы. Они могут появляться как первый тип приступов при развитии заболевания (что бывает значительно редко) или на поздних стадиях заболевания.
Редко бывает, чтобы у пациента были только абсансы или миоклонии, чаще мы сталкиваемся с комбинацией всех трех типов приступов.
Из всех пациентов с генерализованными миоклоническими приступами у 90% лиц наблюдаются тонико-клонические эпиприступы, у 30-40% – абсансные варианты.
Ювенильная миоклоническая эпилепсия обычно хорошо поддается терапевтическому влиянию (Calleja et al., 2001). Однако данный синдром требует долговременного лечения, поскольку, согласно наблюдениям (Delgado-Escueta & Enrile-Bacsal, 1984), около 90% пациентов отмечают возобновление эпиприступов после отмены терапии ПЭП.
В типичных случаях никаких неврологических или психиатрических отклонений не обнаруживается. Интеллектуальные функции пациента обычно не страдают.
Если клинический диагноз, благодаря анамнезу, типичным приступам и отсутствию неврологичксих органических отклонений, кажется ясным, то нет необходимости в нейровизуализации. Небходимо, чтобы клинический диагноз был подтвержден ЭЭГ. У нелеченых пациентов интериктальная ЭЭГ должна показывать типичную генерализованную спайк-волновую активность при нормальной фоновой активности. Отличительным признаком ЮМЭ, однако, являются полиспайк-волновые паттерны, где за разрядами, состоящими из двух и более спайков, следуют медленные волны высокой амплитуды. Это можно увидеть в интериктальном периоде, но также должно быть представлено в том случае, если во время исследования были миоклонические подергивания. Миоклонии без таких паттернов не совместимы с диагнозом.
Если не обнаружено никаких отклонений в стандартном ЭЭГ, его необходимо повторить после депривации сна, и электроэнцефалография должна проводиться во сне, так как эпилептиформная активность возрастает и при засыпании, и при пробуждении (видео-мониторинг-ЭЭГ). Гипервентиляция должна усиливать спайк-волновую активность во всех случаях идиопатической генерализованной эпилепсии, а фотостимуляция положительна (спайк-волновая активность с клиническими симптомами или без них) почти у 50% пациентов с ЮМЭ. Наиболее важная часть диагноза — тщательный сбор анамнеза заболевания, когда подростки или молодые люди с впервые появившимися приступами специально опрашиваются о миоклонических подергиваниях, их зависимости от недосыпания, связи с пробуждением, и описываются рефлекторные эпилептические признаки. Обычно по крайней мере один из них выявляется или клинически, или при ЭЭГ-исследовании. Если на ЭЭГ выявляются типичные межприступные, а особенно приступные признаки, диагноз становится бесспорным.
Видео-ЭЭГ обычно выявляет билатеральную эпилептиформную активность. N. Usui et al. (2006) отметили, что у 14 (54%) из 26 пациентов с ЮМЭ наблюдались клинические или электроэнцефалографические фокальные черты, или сочетание обоих феноменов.
Один из самых актуальных в настоящее время вопросов эпилептологии — дихотомическое деление эпилепсий на фокальные и генерализованные. Широко известен факт, что фокальные формы эпилепсии нередко маскируют генерализованные формы за счет феномена вторичной билатеральной синхронизации (ВБС) и диффузного распространения эпилептической активности с развитием приступов, которые визуально, по кинематике приступа, могут быть расценены как генерализованные. Это явление широко распространено у пациентов с симптоматическими формами эпилепсии, особенно в младенческом и раннем детском возрасте (фокальные «маски» синдрома Отахара, Веста, Леннокса-Гасто и др.), что послужило обособлению особой группы эпилептических энцефалопатий от генерализованных и фокальных форм в проекте новой классификации эпилепсий и эпилептических синдромов. Симптоматические фокальные формы эпилепсии нередко «маскируются» под идиопатические формы (как фокальные, так и генерализованные), и нередко приступы, по внешним характеристикам напоминающие типичные генерализованные, имеют на самом деле фокальный генез (т.е. возникают за счет явления вторичной билатеральной синхронизации с диффузным распространением эпилептиформной активности). Данный феномен послужил основной для определения понятия «псевдогенерализованные» приступы (Мухин К.Ю. и соавт., 2006). С другой стороны, наблюдается противоположный факт — идиопатические генерализованные эпилепсии в ряде клинических случаев имеют фокальные черты в кинематике приступов и на ЭЭГ, но их фокальный характер исключается при применении комплексного клинико-электро-нейровизуализационного диагностического подхода.
В 2000 г. H. Meencke поднял вопрос о том, что дихотомическое деление эпилепсий на генерализованные и парциальные еще требует доказательств. Из доклада по классификации и терминологии ILAE (2001): «…существующая концепция парциальных и генерализованных эпилепсий и отдельных типов приступов, как результата исключительно локальной дисфункции в одной гемисфере или вовлечения всего головного мозга, логически несостоятельна. В частности, могут иметь место: диффузное повреждение головного мозга, мультифокальные аномалии, билатерально-симметричные локальные аномалии… И, хотя дихотомическое деление эпилептогенеза на парциальную и генерализованную составляющую еще применяется на практике, однако оно не может быть применимо ко всем формам эпилепсии и всем типам приступов…».
В России пилотные исследования в области фокальных черт приступов и форм эпилепсии, традиционно считавшихся первично-генерализованными, проводились под руководством академика В.А. Карлова. В.А. Карлов и В.В. Гнездицкий в 2005 г. опубликовали результаты многолетних исследований, в которых показано очаговое начало абсанса. Локализация эпилептического очага определена в большинстве случаев в префронтальной коре, и показано, что в формировании особого типа эпилептической системы так же играет роль таламус. Генерация спайков в лицевой соматосенсорной коре и последующее их распространение на таламус была показана на генетической модели абсанс эпилепсии у крыс (Polack et al., 2009).
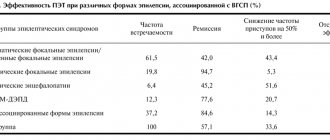
M. Koepp et al. (2005) показали, что при применении различных методов диагностики выявляются признаки очаговой патологии в головном мозге (ПЭТ обнаруживает признаки нейротрансмиттерной дисфункции в коре большого мозга, МР-исследования демонстрируют изменения в коре медиальных отделов лобной доли, при проведении 1H-магнитно-резонансной спектроскопии выявляется дисфункция в таламусе). Все это позволяет предположить, что при ЮМЭ в большей степени, чем при других формах ИГЭ, играет роль вовлечение лобных отделов в структуре эпилептогенных таламокортикальных «сетей», и синдром Янца имеет региональный генез с множественными очагами в лобных отделах.
Имеются сообщения о возможности сочетания двух форм эпилепсии — ИГЭ и фокальной эпилепсии — у одного больного. A. Nicolson (2004) сообщает, что подобный феномен наблюдается менее чем у 1% больных ИГЭ. Zajac et al. (2007) при обследовании с помощью МРТ 45 детей с диагнозом первично-генерализованной эпилепсии в 38% случаев обнаружили фокальные аномалии (кисты, асимметрии желудочков, признаки очаговой демиелинизации, опухоли, глиоз и атрофические процессы). Авторы рекомендуют более тщательный поиск фокального компонента приступов у пациентов данной категории. В литературе немного исследований, касающихся прогноза течения, вариантов исхода ЮМЭ.
Нами проанализированы катамнестические данные 40 больных ЮМЭ, наблюдающихся в клинике неврологии СГМУ и Дорожной клинической больнице г. Саратова в период с 1992 до 2012. Длительность наблюдения от 5 до 20 лет.
Дебют заболевания в пубертатном периоде отмечался у 28 больных(70%). У небольшой группы больных (7 человек, 17,5%) заболевание развилось во взрослом возрасте (18-21 год). Под наблюдением находится 3 больных (12,5%), у которых первый приступ в виде тонико-клонического развился в возрасте 25-30 лет.
Классическое сочетание 3 генерализованных эпилептических приступов в клинической картине заболевания регистрировался нечасто- 11 человек (27,5%). Следует отметить, что абсансы всегда регистрировались при дебюте ЮМЭ в подростковом возрасте и никогда не возникали при дебюте позже. Еще одним важным клиническим аспектом является полная редукция абсансов при переходе пациента во взрослый период. Обычно это происходит на фоне антиэпилептической терапии или спонтанно, в случае более позднего начала терапии.
Сочетание ГТКП и миоклонических составляет наиболее частый вариант приступов при ЮМЭ (25 человек-62,5%). И только у 4 пациентов (10%) ЮМЭ проявлялась только миоклоническими приступами и характерными изменениями ЭЭГ. Изолированный миклонус век в сочетании с ГТКП и ЭЭГ-данными вявлен у 2 больных, а также регистрировался у 4 пациентов в элекроклинической (?) ремиссии ЮМЭ. Адверсивный компонент в начале или при окончании ГТКП отмечен у 15 больных (37,5%), что служило причиной диагностики фокальной эпилепсии у части больных до консультации эпилептолога. В литературе описан феномен инициации приступа как генерализованного с фокальным завершением. Williamson R. et al. (2009), сообщают о 6 пациентах, у которых отмечались приступы с генерализованным началом, далее трансформировавшиеся в фокальные. Приступ начинался с абсанса или миоклоний, после чего могли отмечаться поведенческие нарушения и автоматизмами, и затем возникали постприступные симптомы выпадения (нарушения сознания). На ЭЭГ отмечалась генерализованная активность с дальнейшим появлением региональных нарушений. Интериктальная эпилептиформная активность носила генерализованный характер. При проведении МР-исследований никаких патологических изменений выявлено не было. У 4 пациентов изначально диагностировалась фокальная эпилепсия. При назначении антиэпилептической терапии (АЭП, эффективные в отношении абсансов и миоклоний) у 3 больных приступы купировались полностью, у 3 — частота приступов значительно уменьшилась.
Короткая зрительная аура перед ГТКП в виде «мимолетных» вспышек и «летящих с огромной скоростью солнечных лучей» выявлена нами у 2 больных. В литературе описаны три случая появления зрительных аур непосредственно перед развитием генерализованных тонико-клонических приступов. Результаты исследования показали, что при идиопатической генерализованной эпилепсии возможно возникновение зрительных аур, проявляющихся в виде вспышек света, «молний» или возникающего у больного ощущения, как будто он «видит солнце». В отличие от зрительных аур, описанных при затылочной эпилепсии, зрительные ауры при ИГЭ отличаются очень короткой продолжительностью (Gelisse P. et al., 2008). У 2 пациентов после тяжелого клонико-тонического приступа в раннем постиктальном периоде развивалось психомоторное возбуждение с сумеречным расстройством сознания (фокальный компонент). В литературе часто встречается упоминание о двух разновидностях приступов, которые клинически имеют фокальное проявление — это адверсивные и ротаторные (кручение) приступы. Наиболее часто встречающийся феномен — адверсия головы и глаз (в этих случаях часто ставится диагноз лобной эпилепсии), а в случаях с ротацией — может ставиться диагноз лобной или височной эпилепсии. О подобных феноменах сообщал и H. Gastaut (1986), назвав эту форму заболевания, диагностированную у детей с пик-волновыми разрядами частотой 3 Гц на ЭЭГ, «версивной эпилепсией». Многие пациенты с подобными феноменами имеют, кроме того, типичные абсансы и миоклонические приступы. Получены сообщения о том, что версивные приступы перед развитием ГТКП могут отмечаться в дебюте ИГЭ, причем в дальнейшем направление адверсии или кручения у многих пациентов остается стабильным. Результаты некоторых исследований продемонстрировали отсутствие влияния приступов с адверсией или кручением на прогноз заболевания (Aguglia U. et al., 1999).
ЭЭГ-особенности ЮМЭ
Медленно-волновые изменения, региональные спайки или острые волны, независимые от генерализованных разрядов, региональные спайки, комплексы спайк-волна, медленные волны непосредственно перед генерализованным разрядом выявлены в значительной группе пациентов (17 человек-42,5%). Как правило, они носили характер единичных, коротких, исчезающих и/или вновь появляющихся при динамическом наблюдении. Наиболее часто фокальные компонены ЭЭГ регистрировались при длительной записи ЭЭГ, особенно в период дневного или ночного сна. Отмечено, что наиболее часто эти изменения ЭЭГ регистрировались у больных на фоне лечения, при достижении ремиссии приступов или при развитии фармакорезистентности. В исследованиях C.T. Lombroso (1997) у 32 (56%) из 58 пациентов с ИГЭ наблюдались региональные изменения на ЭЭГ, причем, в дебюте заболевания изменения отмечались лишь у 13% больных. Автор выдвинул гипотезу о том, что у таких больных может иметь место либо независимая кортикальная локальная патология, либо с течением болезни формируется независимый очаг эпилептогенеза. Leutmezer F. et al. (2002), напротив, указывали, что наличие кортикальной аномалии в таких случаях, с большей вероятностью свидетельствует в пользу фокальной эпилепсии. Многие исследователи сообщают об обнаружении региональных изменений на ЭЭГ у 1/5–1/2 пациентов с ИГЭ (Panayiotopoulos C.P. et al., 1991; Montalenti E. et al., 2001; Aliberti V. et al., 1994; Lombroso C.T., 1997).
МРТ картина у большинства больных не выявляла органических изменений в головном мозге (28 человек-70%). В остальных случаях (30%) на МРТ выявлялись диффузное расширение субарахноидальных пространств, сообщающуюся гидроцефалию,внутреннюю симметричную гидроцефалию, ретроцеребеллярные кисты.
Монотерапия проводилась у 26 пациентов (65%), дуотерапия у 12 человек (35%) препаратами выбора являлись Вальпроаты, с эффектом применялись Топирампат, Кеппра, Барбитураты. В единичных наблюдениях, особенно при фотосенситивности, применялиЛамотрижин (учитывалась вероятность аггравации приступов при ЮМЭ). Стойкая электроклиническая ремиссия отмечена у 31 больного (77,5%). Многолетняя ремиссия приступов, с регистрацией эпилептиформных изменений, что не позволяло в ряде случаев отменить антиконвульсанты, отмечена в 5 случаях. К сожалению, у 4 (10%) больных отмечалось фармакорезистентное течение заболевания, что не позволило достичь многолетней ремиссии приступов. В лучшем случае, межприступные интервалы достигали 8-10 месяцев.
Тщательного и взвешенного подхода требует факт появления эпилептиформной активности на ЭЭГ у больных в ремиссии ЮМЭ. Наши наблюдения показывают, что в случае регистрации фокальных спайков, особенно во сне, можно воздержаться от немедленного возврата к назначению антиконвульсантов без высокого риска. Конечно, при этом необходим более частый контроль ЭЭГ-мониторинга (через 3,6 месяцев).
срывы ремиссий.
Эффективность терапии
ЮМЭ характеризуется в ряде случаев высокой чувствительностью к антиконвульсантам. Степень достижения стойкой электроклинической ремиссии составляет 80% (32 человека). У 20% больных не удалось достичь ремиссии приступов более чем на 1-2 года, в первую очередь это касается рецидивов миоклонических приступов. Как правило, миоклонии являлись спровоцированными (депривация сна, стрессовые факторы, прием алкоголя, перерыв в приема антиконвульсантов на 1-2 суток и др). Несомненным является факт более длительного срока приема антиконвульсантов при ЮМЭ. Чем длительнее срок приема препаратов, тем меньше вероятность редицива приступов. Как правило, сроки терапии при достижении электроклинической ремиссии при ЮМЭ достигали не менее 5-6 лет. При сроке ремиссии 3-3,5 года вероятность редицивов после отмены препаратов возрастает. Из 12 больных со сроками ремиссии 3-3,5 года у 8 (66,6%) в течение первого года после отмены антиконвульсантов либо рецидивировали миоклонические или ГТКП, либо появлялась генерализованная спайк-волновая активность при фотостимуляции или во сне. Появление такой активности расценивали предиктором высокого риска развития приступов и вновь назначали антиконвульсанты в прежних дозировках. В общей сложности, 12 больных (30%) в группе ЮМЭ длительно получали антиконвульсанты (от 5 до 10 лет). Как указывалось выше,4 больных явились фармакорезистентными. Т.е. группа больных ЮМЭ длительно получающих терапию, составила 16 человек (40% ), в силу развития рецидивив приступов, появления спайк-волновых феноменов на ЭЭГ или фармакорезистентности.
На наш взгляд, паттернами высокого риска срыва ремиссии ЮМЭ являются:
- Появление фотосенситивных разрядов на ЭЭГ, особенно в стадии клинической ремиссии
- «Плохой» ответ ГТКП на антиконвульсанты
- Политерапия ЮМЭ
- Наличие у пациента клонико-тонико-клонических судорожных приступов ( тонико-клонических судорог с предшествующими массивными билатеральными миоклоническими судорогами).
Атипичный вариант течения ЮМЭ у взрослых
Ранее нами (Коротков А.Г., Кабанова Л.А.,2006) описаны 2 случая доброкачественного течения идиопатической генерализованной эпилепсии у взрослых, соответствующих критериям ЮМЭ. Особенность обоих случаев- длительный анамнез заболевания с редкими миоклоническими приступами и единичным ГТКП без назначания антиконвульсантов. Лишь появление повторного ГТКП ( в 1 случае-через 15 лет от начала заболевания, во 2 наблюдении- через 18 лет повтор в виде клонико-тонико-клонического приступа) заставило пациентов обратиться в неврологу и провести соответствующее обследование. Катамнестически удалось выяснить, что периодически, но нечаще 1-2 раз в год у больных возникали утренние миоклонии, чаще провоцированные депривацией сна. На ЭЭГ-мониторинге дневного сна в обоих случаях выявлена генерализованная спайк волновая активность, на МРТ головного мозга-без органических изменений. Под наблюдением находится еще 1 пациентка 36 лет, у которой с 18 лет наблюдаются периоды серий миоклоний в руках, иногда с вовлечением ног, с периодичностью 1 в 1-2 года. Работает медицинской сестрой, по роду профессиональной деятельности, работа связана с депривацией сна. Серии миклонии до 3-4 минут возникали обычно после ночных дежурств в утренние часы. Неоднократно обследована по этому поводу (рутинная ЭЭГ, МРТ), которые не выявляли патологию и подергивания в руках расценивались неврологом как конверсионное расстройство.. В 2012 году наблюдались 2 серии миоклоний после депривации сна, впервые произведен мониторинг ночного сна, выявлена генерализованная полиспайк-волновая активность. На наш взгляд, у пациентки ЮМЭ в доброкачественным течение, не требующим назначания антиконвульсантов в настоящее время.
Таким образом длительное катамнестическое наблюдение (от 5 до 20 лет) за 40 больными ЮМЭ позволило прийти к следующим заключениям:
1. ЮМЭ является неоднородным по клиническим проявлениям синдромом генерализованной эпилепсии. В структуре приступов ЮМЭ в ряде случаев имеются фокальные симптомы.
2. В подавляющем большинстве (60%) случаев при ЮМЭ у взрослых удается достичь многолетней клинической ремиссии и отменить антиконвульсанты.
3. В группе ЮМЭ 40% больных нуждаютя в многолетней терапии из-за фармакорезистентности, рецидивов приступов или появления спайк-волновой активности на ЭЭГ после отмены препаратов.
4. Появление единичных, коротких билатеральных (бифронтальных) спайк-волновых разрядов в ремиссии заболевания после отмены антиконвульсантов не всегда требует однозначного возврата к назначению препаратов.
5. По-видимому, существует вариант доброкачественного (атипичного?) течения ЮМЭ у взрослых с единичным ГТКП и крайне редкими редицивирующими провоцированными миоклоническим приступами, не требующий назначения антиконвульсантов.
Течение и лечение
У детей с СДМЭД не наблюдаются припадки какого-либо другого типа, даже если они остаются без лечения (до 8,5 лет у одного нашего больного), особенно это касается малых эпилептических или тонических припадков. Результаты клинического обследования нормальные. Интериктальный миоклонус был описан только Джиованарди-Росси и др. (1997) у 6 больных. Анализируя состояние наших больных, мы обнаружили легкий интериктальный миоклонус у 2 по записям ЭЭГ. Многие больные не были обследованы, но когда КТ и МРТ были выполнены, результаты оказались нормальными (33 больных).
Исход, вероятно, зависит от раннего диагноза и лечения. Миоклонии легко поддаются монотерапии вальпроатом, развитие ребенка при этом соответствует возрасту. При отсутствии лечения у больного продолжаются миоклонические припадки, что может привести к нарушению психомоторного развития и поведенческим отклонениям.
Более или менее досконально способы лечения апробированы на 74 больных. У 65 больных проводилась монотерапия, у 6 — политерапия, у 3 лечение не проводилось. Монотерапия включала вальпроат (ВПА), фенобарбитал (ФБ), нитразепам (НТЗ). 6 больных получали терапию примидоном (ПРМ) и этосуксимидом (ЭСМ). В результате лечения припадки исчезли у 69 больных (93%).
Эти данные подтверждают, что при СДМЭД ВПА является препаратом первого выбора. Однако лечение должно проходить под контролем концентрации препарата в плазме, т.к. неправильный прием может привести к рецидиву или вызвать резистентную к препаратам эпилепсию.
Отдаленные результаты и прогноз
Длительность наблюдения за 63 больными составила от 9 мес. до 27 лет, из них за 45 пациентами наблюдали около 5 лет. Возраст больных в период наблюдения — от младше 5 до старше 15 лет.
Во всех случаях МП купировались. Длительность заболевания известна у 52 больных: у большинства из них МП длились менее одного года, у 7 — от 1 до 2 лет, только у 5 — более двух лет.
О возникновении генерализованных эпилептических припадков (ГЭП) после прекращения МП сообщалось в случае 10 больных без сопутствующих МП.
Было доложено о результатах наблюдения 74 больных.
Приступы прекратились у 28 больных в возрасте старше 6 лет. У 6 детей этого же возраста приступы сохраняются: при фотостимуляции — у 2 (Драве и др., 1992), по неизвестной причине — у 3 (Джиованарди-Росси и др., 1997). Также приступы сохраняются у 13 больных младше 6 лет. 3 больных младше 6 лет остались без лечения (Риччи и др., 1995). О 24 больных информации нет.
Картина ЭЭГ известна для 55 больных. Она быстро нормализовалась у 23. Редкие спонтанные генерализованные СВ сохранялись у 13. Фотосенситивные изменения наблюдались у 6. Интересно, что фотосенситивность может появиться после исчезновения МП и персистировать долгие годы после прекращения МП вплоть до взрослого возраста. Были также представлены фокальные нарушения. Они были записаны у 5 больных во время пробуждения как СВ в двух лобно-центральных и теменных областях, иногда в лобно-теменной и лобно-височной. Они исчезали с течением времени. Напротив, у других больных они появлялись только во время сна. Они еще сохранялись в конце периода наблюдения у 5 наших больных, и только во время сна.
В целом психологический исход благоприятный, и большинство больных выздоровели. Это точно известно в 69 случаях. 57 (83%) были здоровыми, из них 38 в возрасте 5 лет и старше. У 10 (14%) была легкая заторможенность, они посещали специализированную школу, но никто из них не был помещен в больницу. У 2 больных (3%) была нарушена познавательная способность и выявлены отклонения в формировании личности: у одного пациента был синдром Дауна и сильная чувствительность, у другого до 5 лет были МП с чувствительностью к ИРС и к закрытию глаза. Это психотическое нарушение появилось у него в возрасте 10 лет и прогрессировало.
Этот психологический исход частично зависит от ранней диагностики, позволяющей провести соответствующее лечение и убедить семью в хорошем будущем.
Эти данные подтверждают обычно хороший прогноз СДМЭД. Приступы, вызванные шумом или разговором, было легче контролировать, чем спонтанные. Фотосенситивность же было труднее контролировать, она сохранялась на протяжении нескольких лет после прекращения припадков.
Прогноз
Юношеская миоклоническая эпилепсия считается хроническим заболеванием, которое продолжается в течение всей жизни пациента. Случаи спонтанной ремиссии редки. У 90% больных, по различным причинам прервавших лечение антиэпилептическим препаратом, отмечалось возобновление эпиприступов. Однако имеются указания на то, что в отдельных случаях у пациентов после отмены препарата наблюдалась длительная ремиссия.
В целом, при правильно подобранной терапии приступы контролируются у большинства больных, хотя у половины из них на фоне лечения могут наблюдаться отдельные пароксизмы. Относительно резистентное к терапии течение наблюдается редко, в основном в случаях, когда у пациента отмечаются все 3 разновидности пароксизмов ЮМЭ.
Вы можете поделиться своей историей болезни, что Вам помогло при лечении юношеской миоклонической эпилепсии.
Дифференциальный диагноз
Если миоклонии начинаются в первый год жизни, их надо дифференцировать с криптогенными детскими судорогами (ДС). ДС клинически отличаются от доброкачественных миоклоний: они более тяжело протекают, включают в себя тонические судороги во всем теле, что никогда не наблюдается при СДМЭД; единичные судороги всегда сочетаются с серийными приступами у данного ребенка; возникновение долгих серийных спазмов происходит после пробуждения. Затем СД на ЭЭГ демонстрируют типичную картину коротких тонических сокращений, которую хорошо описали Руско и Вигевано (1993); редко имеют место длительные миоклонии. Иктальная ЭЭГ переменная: внезапные прерывистые гипсаритмии выравниваются наложенными быстрыми ритмами, большой медленной волной, за которой следует выравнивание или отсутствие видимых изменений. Наступление ДС связано с поведенческими изменениями, затруднением речевого контакта, замедлением психомоторных достижений.
На интериктальной ЭЭГ всегда есть патологические признаки: истинная гипсаритмия, или видоизмененная гипсаритмия, или фокальные нарушения, на них не определяются отдельные или короткие всплески билатеральных синхронных СВ или СДМЭД.
Если психомоторное развитие и ЭЭГ в пределах нормы по результатам нескольких обследований, выполненных во время бодрствования и сна, припадки, похожие на ДС, должны навести на мысль о диагнозе доброкачественного неэпилептического миоклонуса, описанного Ломброзо и Фейерманом (1977). У этих больных даже иктальная ЭЭГ без патологических изменений (Драве и др., 1986; Пашатс и др., 1999).
На первом году жизни может развиваться миоклоническая эпилепсия младенчества, она всегда начинается с длительных и частых фебрильных судорожных припадков, а не с отдельных миоклонических приступов. На втором году психомоторное развитие замедляется.
Если миоклонии начинаются после первого года жизни, можно заподозрить криптогенный синдром Леннокса — Гасто (СЛГ). При СЛГ (Бюмане и Драве, 1992) приступы в основном не миоклонические, а миоклонико-атонические, а чаще тонические, приводящие к внезапным падениям и травмам. Их ЭЭГ картина неоднородна, а в иктальном периоде высокоамплитудная или выравнивается, или определяется высокая медленноволновая активность, за которой следуют низкоамплитудные волны. В самом начале интериктальные ЭЭГ могут быть нормальными, в дальнейшем могут появиться типичные диффузные разряды медленной СВ. Типичные электроклинические признаки во время сна могут быть отложенными по времени. Диагноз основывается на быстрой связи припадков разного типа, таких как атипичные абсансы и аксиальные тонические судороги, на постоянных поведенческих нарушениях поведения и появлении новых навыков и неэффективности антиэпилептических препаратов.
Если миоклонические приступы изолированные или связаны с БЭП, необходимо рассмотреть диагноз миоклонической астазической эпилепсии раннего детского возраста (ЭМАП), хотя при этом синдроме миоклонические астанические припадки редко начинаются до возраста 3 лет (Дуз, 1992). Есть два существенных различия: 1) клинический аспект припадков со ступором, которые не наблюдаются при СДМЭД (Гуеррини и др., 1994); 2) ЭЭГ признаки тоже разные: СВ и ПСВ более многочисленные, сгруппированы в долгие вспышки, связанные типичным тета-ритмом над центро-теменными зонами. Но некоторых больных, вероятно, следовало бы классифицировать как СДМЭД. Подобным образом Дельгадо-Эскуэта и др. (1990) в группу обследованных или больных включили, вероятно, под названием миоклонической эпилепсии детского возраста (МЭРД) случаи как ЭМАП, так и СДМЭД.
Наконец, следует учитывать и другие эпилепсии, начинающиеся в первые три года жизни, при которых миоклонии являются основным типом приступов и которые имеют изменчивый прогноз. Они неоднородны: сочетание приступов других типов, постоянные фокальные нарушения на ЭЭГ, замедленное психомоторное развитие в прошлом, плохая реакция на лечение, неясный прогноз (Драве и др., 1992).
Диагностика
Подтвердить наличие юношеской миоклонической эпилепсии на начальной стадии ее развития очень сложно. Часто проявление симптомов ЮМЭ в раннем возрасте списываются на повышенную возбудимость, нервозность ребенка, сами же дети проявления симптомов практически не замечают.
В первую очередь для постановки диагноза необходимо исключить возможность наличия у ребенка других видов церебральных патологий (рака, абсцесса мозга, внутримозговой кисты, отека и наличие вирусного и бактериального поражения мозга). Данный этап диагностики не только жизненно важен, но и крайне сложен, так как симптомы патологий данной группы практически идентичны. Для подтверждения миоклонической формы применяется МРТ, ангиография.
Дальнейшая диагностика проводится под руководством эпилептолога. Под его руководством проводится электроэнцефалография, которая практически в 70% случаев позволяет характерные для юношеской миоклонической эпилепсии симптомы.
Кроме электроэнцефалограммы для ранней диагностики применяются ЭЭГ исследование (при пробуждении, а также засыпании), суточный мониторинг ЭЭГ, депривация сна, фотостимуляция.
Дифференциальная диагностика
ЮМЭ требует четкой дифференциации от других видов эпилепсии, обладающих сходными симптомами. Свойственные для ЮМЭ пароксизмы от пароксизмов свойственных патологиям другой этимологии отличает синхронность приступов. Еще один важный диагностический критерий —наличие миоклонических приступов, которые при ЮМЭ практически не встречаются.
Также практически все виды патологий нервной системы, манифестирующие в раннем возрасте, сопровождаются серьезными задержками психического, умственного развития, которые при наличии миоклонической эпилепсии не встречаются.
Кроме того, ЮМЭ практически никогда не осложняется приступами, при которых судороги дополняются абсансами и пароксизмами, протекающими без потери сознания.
Клиническое обследование больного с целью постановки диагноза.
Это обследование — достаточно простое. Для него необходим тщательный сбор анамнеза и повторные видео-ЭЭГ записи, чтобы продемонстрировать наличие МП с генерализованными СВ разрядами, спонтанными или такими, которым способствуют сонливость, шум, контакт или ИРС. Записи сна позволяют показать слабую активацию разрядов и фокальных нарушений. Полезно использовать нейровизуализацию для подтверждения отсутствия поражения мозга, но она не является обязательной при наличии типичной симптоматики. Более полезно психоневрологическое обследование для проверки психомоторного развития и прослеживания течения заболевания.
Лечение
ВПА в монотерапии является препаратом выбора, его применение следует начинать как можно раньше. Предпочтительнее применять раствор, а не сироп, так как он лучше переносится ребенком. Необходимо строго следить за концентрацией препарата в плазме.
Обычно бывает достаточно использовать дневную дозу 30 мг/кг 3 раза / день, но для некоторых больных может потребоваться и более высокая доза. ВПА также эффективен при возможных фебрильных припадках. Если ВПА дает неполный эффект, можно использовать его в сочетании с бензоадиазепином (СLВ или НТЗ) или ETS и пересмотреть диагноз. Лечение следует проводить в течение 3-4 лет после начала заболевания, если оно хорошо переносится. В случаях чисто рефлекторных признаков медикаментозную терапию можно не применять. Если она уже начата, ее можно резко прекратить, однако если отсутствует сверхчувствительность. Если БЭП случается у подростка, можно провести кратковременное лечение в этом возрасте. Консервативное лечение должно сочетаться с психологической помощью семье.