- Home >
- Directory >
- Why is the sella turcica empty?
Some patients suffering from headaches, dizziness, increased fatigue, and obesity cannot always find the causes of their suffering.
Even doctors with a traditional approach to examination are unable to identify the source of the disease. And if a patient, for example, complains of a decrease in the quality of vision, performance, or short stature (nothing helps him grow), then in the best case he will be referred to an endocrinologist or ophthalmologist, in the worst case he will be advised to vitaminize the body and take a break from work in a sanatorium. everyday life.
But if the “root of evil” lies precisely in workload, lack of sleep or lack of microelements and vitamins, then such advice is quite realistic and can be beneficial. If there are more serious problems, some kind of pathology occurring in the brain, this restorative therapy will only have a temporary effect. This will happen due to the fact that the patient will calm down and will pay less attention to the symptoms, believing that it is all a matter of fatigue and a seasonal lack of useful energy substances.
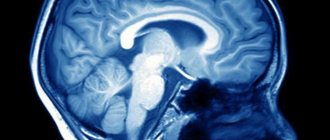
And only by chance, when examining the brain for the presence of tumors, aneurysms, sinusitis or consequences of trauma, is an empty sella turcica discovered on MRI (if such a syndrome exists, but it may not exist). Of course, this is not the main indicator of the source of the ailment, but such a diagnosis makes it possible to associate cause and effect, for example, dizziness, blurred vision, or discharge of CSF (cerebrospinal fluid) through the nasopharynx.
Where is the sella turcica located?
Patients who, after magnetic resonance imaging, are told that an “empty sella turcica” has been found in the brain, begin to panic because they have no idea what they are talking about. And the word “empty” generally confuses some people, since they associate this with the lack of some tissues necessary for their body. When the doctor explains that there are no serious problems, they do not believe it, believing that the truth is being hidden from them. Although, if the doctor does not see the reasons for the unrest, you should not distrust him.
This anatomical organ of the brain appendage is “responsible” for the production of hormones that affect human growth, reproduction, and metabolism. If this same pituitary gland does not completely fill the cavity of the sella turcica, then the free space is “closed” by the meninges, i.e. cerebrospinal fluid (CSF). When examined on a magnetic resonance imaging scanner, the thinnest section shows in this case an increase in the size of the “sella turcica”. It is precisely this anatomical condition of this part of the brain structure that is called the “empty sella” syndrome.
Diagnostics
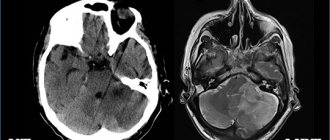
To diagnose SPTS, the doctor uses instrumental examination methods, such as MRI and CT. The image will show any pathological changes in the brain tissue, including the pituitary gland.
In more than half of the cases, the sella turcica is detected by chance, for example, when a full diagnosis is prescribed using magnetic resonance imaging. The disease often manifests itself insignificantly, so a person does not even feel much discomfort; it is especially difficult to find out about its presence against the background of heavy physical labor. The problem is often detected when signs of diabetes occur, as well as when the thyroid gland is abnormally enlarged and vision deteriorates.
For a comprehensive diagnosis, you will need to take a blood test to see if there are hormonal imbalances in the body. Such an examination will take a little more time, but you will be able to accurately verify your diagnosis and safely begin a course of treatment.
Empty sella syndrome
Primary syndrome
According to medical statistics, it is more common, as it is associated with a congenital anomaly of the sella diaphragm, due to which CSF easily penetrates into the sella turcica. This pathology is diagnosed during an MRI examination in 10%-40% of people who do not complain about their health, because the work of the pituitary gland in this case, as a rule, is not impaired. But as a result of changes in the pressure of the cerebrospinal fluid on the pituitary gland, the sella turcica increases in size, which is recorded on a magnetic tomograph. Typically, primary empty sella syndrome is diagnosed in 30-40 year old women who have problems with obesity.
Secondary syndrome
The reasons for the “emptying” of the sella turcica are a significant reduction in the size of the pituitary gland or the destruction of enlarged tumor tissue as a result of treatment or surgery. Hemorrhage of the pituitary gland into the tumor can also occur and, as a result, the cavity of the sella turcica expands significantly and thus the empty sella syndrome is noted.
Causes
Acquired syndrome can be a consequence of a large number of diseases. So, it can be influenced by:
- hormonal changes during puberty, pregnancy, its interruption, menopause, surgical removal of the ovaries, gender reassignment;
- treatment with hormonal drugs, including the use of contraception;
- cardiovascular and neurological disorders (high blood pressure, poor circulation, brain tumors, heart failure, pituitary cysts and adenomas, hypoxia of brain cells, stroke);
- various inflammatory processes, especially those caused by viral infections and those in which long-term antibiotic treatment was used;
- autoimmune diseases;
- excess weight;
- traumatic brain injuries;
- surgical measures on the pituitary gland;
- treating cancer with radiation or chemotherapy.
Most often, the disease appears in women aged 35-55 years. Of these, those who have carried more than two children are at risk. The emerging syndrome is often provoked by obesity: more than two thirds of all patients are overweight.
Diagnosis and treatment
As mentioned earlier, this syndrome is not specifically diagnosed. This MRI phenomenon is detected only during brain tomography if there is a suspicion of pituitary tumors or the causes of headaches, dizziness, leakage of cerebrospinal fluid through the nose, consequences of injuries and other ailments are sought.
When scanning the tissues inside the skull, the CSF is noted on the images in the recess of the sella, while the pituitary gland is slightly pushed back to some wall (posterior or lower) of the cavity. And only when more serious pathologies are detected in this anatomical area of the head, an additional expanded diagnosis of the syndrome (primary or secondary - it doesn’t matter) of the empty sella turcica is carried out.
Treatment for such a diagnosis (such as an MRI phenomenon) is usually not required if there are no obvious pathological processes affecting well-being.
Sign up at the MART medical center in St. Petersburg (see map) by calling 8 or leave a request on the website.
Treatment of the disease
First of all, the patient should be referred to a therapist, who, based on a survey and examination of the patient, will redirect to a more specialized specialist. Most often, this is a neurologist and he will prescribe an examination to differentiate empty sella syndrome, determine its type (congenital or acquired) and prescribe treatment
If a person has had the pathology since birth, then usually no special action is required, but the following rules should be followed:
- Play sports, but preferably without much physical stress;
- Correctly plan your diet so that the body receives all the substances it needs and limit the consumption of unhealthy foods, for example, sweets, fast food, as well as fatty and smoked foods;
- Once a year, be examined in a hospital and be registered with a doctor.
If symptoms of the disease are present, the specialist will prescribe medications to relieve migraines, improve the body’s natural resistance, normalize the menstrual cycle, etc. The course is selected strictly individually and it is prohibited to change the dosage on your own.
In the case of acquired syndrome, hormonal therapy is prescribed to normalize the functions of the endocrine system. In rare cases, surgery is used, but mainly due to the development of blindness. The essence of the operation is to correct the membrane and remove tumors, if any.
The course of treatment is usually done without traditional medicine methods, since they cannot help in any way. Instead of unconventional methods of therapy, doctors advise getting rid of excess body weight and stopping using hormonal medications. It is also advisable to give up bad habits and eat right.
Sella turcica is a fairly common brain disease. It often does not manifest itself, but if symptoms are present, then treatment consists of stopping attacks and a healthy lifestyle. In severe situations, surgery to restore the membrane cannot be avoided.
Forecast
- Sometimes treatment of the underlying disease against which the syndrome appeared eliminates its manifestation.
- With drug adjustment of the amount of hormones in the blood, dysfunction may not appear.
- After surgery, it is impossible to predict the further course of the disease. Forecasts are contradictory and depend on the course of the disease, the condition of the gland, brain and concomitant diseases.
Engage in general health promotion, prevention of injuries, both head and intrauterine. This will help your body fight the disease.
15.09.2016
International Neurological Journal 3 (33) 2010
The term “empty sella syndrome” (PTS) should be understood as prolapse of the suprasellar cistern into the cavity of the sella turcica [1, 2]. The entrance to the sella turcica is normally covered by the dura mater, called the sella diaphragm. The diaphragm separates the cavity of the sella turcica and the pituitary gland from the subarachnoid space, excluding only the hole through which the pituitary stalk passes. The attachment of the diaphragm, its thickness, and the nature of the opening may have significant anatomical variations. Insufficiency of the diaphragm is a prerequisite for the formation of PTS [2–4]. The term “empty sella turcica” should not be taken literally: it is filled with cerebrospinal fluid, pituitary tissue, and sometimes the chiasm and optic nerves can “sag” into it [1–4]. Clinical symptoms associated with an empty sella are accompanied by cephalgia, neuroendocrine and visual disorders [1, 2, 4, 5]. However, according to the literature, from 10 to 50% of congenital defects of the diaphragm sellae may remain clinically asymptomatic for a certain time [1–3, 6].
The phrase “empty sella turcica” was introduced into medicine by W. Busch in 1951. He was the first to associate a partially empty sella turcica with diaphragmatic insufficiency [2, 6].
There are primary (due to congenital or acquired insufficiency of the diaphragm sellae) and secondary PTS syndrome (developing after surgery or radiation therapy for a pituitary tumor) [2]. It has been established that two conditions are necessary for the formation of PTS: diaphragmatic insufficiency and intracranial hypertension [1, 2, 4, 7].
Factors contributing to the development of an empty sella turcica:
- hereditary inferiority of connective tissue;
- increased intracranial pressure (due to pulmonary heart failure, arterial hypertension, traumatic brain injury, brain tumors, sinus thrombosis);
- spontaneous necrosis of pituitary adenoma, pituitary infarction;
— infectious diseases with a severe course (meningitis, hemorrhagic fever, arachnoid cysts that developed as a result of optochiasmal arachnoiditis);
- autoimmune diseases (autoimmune thyroiditis, Sjogren's disease, lymphocytic adenohypophysitis);
- primary hypofunction of peripheral endocrine glands, long-term use of oral contraceptives;
— physiological processes (pregnancy, childbirth, menopause).
There are PTS syndrome without clinical manifestations (PTS symptom) and PTS syndrome with hypothalamic-pituitary and visual disturbances [1, 2, 4, 7]. Reducing the size of the pituitary gland does not always lead to a decrease in its secreting ability. Hypopituitarism is observed only when damage is 70–75%, and panhypopituitarism is observed when at least 90% of the pituitary gland is damaged [8]. In primary empty sella syndrome, the function of the pituitary gland is usually not impaired.
The clinical picture is characterized by dynamism, the replacement of one syndrome by another, and spontaneous remissions. Women are more often affected [1, 2, 5]. The most common symptom is chronic headache (80–90%), which does not have a clear localization and varies from mild to unbearable. About 75% of patients are obese. Dysfunction of the hypothalamus is expressed by vegetative syndromes and vegetative crises with chills, a sharp rise in blood pressure, cardialgia, shortness of breath, a feeling of fear, pain in the abdomen, in the extremities, and fever. Syncope is often observed. Along with autonomic disorders, patients with PTS syndrome are susceptible to emotional, personal and motivational disorders. Clinical symptoms and the course of the disease are aggravated due to acute or chronic stressful situations [1, 2, 5, 9–11].
Endocrine symptoms in PTS are caused by a violation of the tropic function of the pituitary gland, manifest themselves in the form of hypo- or hypersecretion and vary in severity from subclinical to severe forms [2, 8, 10, 12]. The most characteristic manifestations are hypothyroidism, hyperprolactinemia and sexual disorders (decreased potency, libido, oligo- and amenorrhea), Ishchenko-Cushing disease, acromegaly [2, 13–15]. The cause of endocrine disorders in PTS is considered to be not compression of the secretory cells of the pituitary gland, which continue to function even with significant hypoplasia, but a violation of hypothalamic control over the pituitary gland as a result of difficulty in the supply of neurohormones from the hypothalamus [1, 2, 4, 15, 16].
Ophthalmological examination in patients with PTS syndrome is of utmost importance for diagnosing the disease and choosing treatment tactics. The threat of vision loss is an indication for surgical intervention [6, 18, 19]. Changes in the visual system vary in nature and severity [2, 7, 10, 17, 18]. Most often, patients are bothered by retrobulbar pain, accompanied by lacrimation, chemosis, diplopia, photopsia, and blurred vision. Reduced acuity and changes in visual fields, edema and hyperemia of the optic disc, detected during examination, are subject to fluctuations and depend on liquor distension and blood supply to the chiasmatic-optic tract [10, 19]. PTS syndrome is characterized by visual field defects. Two pathogenetic concepts of visual field impairment in PTS have been formed: traction and ischemic. According to the first, defects in the visual fields can be caused by tension between the chiasm and the anterior edge of the diaphragm (when the latter is displaced into the cavity of the sella turcica), as well as tension between the chiasm and the pituitary stalk (when the stalk is displaced back and to the side). The second is considered to be compression of the ophthalmic artery in the subarachnoid space surrounding the optic nerve, and deterioration of the blood supply to the chiasm itself and the optic nerve [2, 17]. Bitemporal hemianopsia, central and paracentral scotomas are more common, and quadrant and binasal hemianopsia are less common [2, 9, 17–19].
The sensitivity of MRI in diagnosing PTS syndrome is almost 100% [3, 20, 21]. PTS is characterized by a triad of symptoms:
1) the presence of cerebrospinal fluid (CSF) in the cavity of the sella turcica, as evidenced by zones of a homogeneous low-intensity signal in the T1W mode and a high-intensity signal in the T2W mode, the pituitary gland is deformed, has the shape of a sickle or crescent up to 2–4 mm thick, its tissue is isointense white matter of the brain, the infundibulum is usually located centrally;
2) asymmetric prolapse of the suprasellar cistern into the sellar cavity, displacement of the infundibulum anteriorly, posteriorly or laterally;
3) thinning and lengthening of the pituitary funnel.
In addition to the main changes in the parasellar region, MRI allows us to identify indirect signs of intracranial hypertension (dilation of the ventricles and liquor-containing spaces), which accompanies this pathology.
Laboratory diagnosis is based on the determination of tropic hormones of the pituitary gland, however, in PTS syndrome there is no certainty and stability in these indicators [1, 5, 12].
Primary PTS syndrome usually does not require treatment. Occasionally, there is a need for hormonal replacement and symptomatic therapy. On the contrary, in secondary PTS syndrome, hormone replacement therapy is almost always necessary. Very rarely, nasal leakage of CSF is observed, which leaks through the thinned bottom of the sella turcica, which also serves as an indication for surgery for primary empty sella syndrome.
The presence of epileptic seizures, mild cognitive impairment, signs of mesoectodermal dysplasia [11], data from neurophysiological, neuroophthalmological, radiological and neuroimaging studies contributed to the search for syndromes associated with gene mutations encoding primary PTS syndrome, as well as with pathology of other organs and systems, the description of which is given below.
605020 (MIM) Craniofacial abnormalities including empty sella turcica, corneal endothelial changes, and retinal and auditory bipolar cell abnormalities. The gene is mapped to 20p11.2 and named visual system homeobox gene 1 (VSX1). VSX1 has been identified in embryonic craniofacial structures, embryonic and adult brains, heart, kidneys, liver, lungs, skeletal muscles, spleen and thymus [22], and in the adult retina, where it controls the distribution of red-green visual pigment [23]. Heon et al. (2002) identified a VSX1 gene mutation in 2 inherited human corneal dystrophies: posterior polymorphous corneal dystrophy (PPCD1) and keratoconus [24]. However, with any VSX1 mutation, retinal dysfunction was detected electroretinographically. Mintz-Hittner et al. (2004) found a GT transversion in codon 256 of the VSX1 protein, which leads to the exchange of alanine for serine (A256S) [25]. Clinical findings demonstrated extremely variable expressivity, but all individuals had wide interpupillary distance, abnormal corneal endothelium, and unusual pinna shape. Other variations included a partially or completely empty sella turcica, sometimes a posterior fossa cyst or an anterior encephalocele and/or hydrocephalus. Electrophysiological studies have provided evidence of abnormalities of retinal bipolar cells (on visual evoked potentials and electroretinogram) and auditory bipolar cells (on audiogram and brainstem auditory evoked potentials) in prospective studies in adults.
612913 (MIM) Orofaciodigital syndrome XI; OFDS XI; Gabrielli's syndrome. Spontaneous mutation, isolated cases.
Gabrielli et al (1994) reported a new OFD syndrome associated with skeletal and craniofacial abnormalities with severe psychomotor retardation [26]. A CT scan of the skull showed duplication of the vomer, clefting of the ethmoid bone, incomplete apophysis of the crista galli, dehiscence of the ethmoidal plate and cleft palate, hypoplastic and deformed dental processes. Nonfusion of the posterior arch of C2 and C3, partial synostosis between the atlas and occipital bone, and clefting of the cervical vertebral bodies were also observed. Ferrero et al. (2002) described a girl with OFD syndrome associated with a nasopharyngeal polyp, partial synostosis between the atlas and occipital bone, multiple proximal-cervical and distal-thoracic vertebral clefts, kyphoscoliosis, empty sella and cognitive impairment [27].
182230 Septooptic dysplasia. De Morsier syndrome. Septoptic dysplasia (SOD); the gene is mapped to 3p21.2-p21.1; HESX1 mutation. Septo-optic dysplasia is a clinically heterogeneous disorder that is defined by any combination of optic nerve hypoplasia, pituitary hypoplasia, and midline brain abnormality, including absence of the corpus callosum and septum pellucidum [28]. Forms with cardiomyopathy have been described. Willnow et al (1996) reported data from a study of 18 patients with SOD. CT or MRI showed the following results: 4 patients had a septum pellucidum defect, 3 had cerebellar hypoplasia, 1 had aplasia of the corpus callosum, and 1 had aplasia of the falx. An empty sella turcica without displacement or with displacement of the pituitary gland was observed in 4 cases. Severe psychomotor retardation was present in 14 of 18 patients. All patients were short. Head circumference and weight were normal. A high prevalence of pituitary dysfunction has been identified, usually with deficiency of somatotropic and thyroid-stimulating hormones [29].
222448 Faciooculoacousticorenal syndrome. Donnai-Barrow syndrome/faciooculoacousticorenal syndrome (DBS/FOAR syndrome). Autosomal recessive. The gene is mapped 2q24-q31, mutation in LRP2.
Manifestations of the clinical picture are polymorphic [30]: macrocephaly, enlarged fontanel, widening of the metopic suture, hypoplasia of the facial skull. Low-lying and posteriorly rotated ears, sensorineural hearing loss. Short, splayed nose. From the organ of vision: hypertelorism, high myopia, coloboma and hypoplasia of the iris, cataracts, Down-like eyes, supraorbital skin folds, retinal dystrophy, exophthalmos. Cardiovascular defects: often septal ventricular defect, rarely duplication of the ascending part of the vena cava. Respiratory abnormalities: pulmonary hypoplasia, secondary diaphragmatic hernia, diaphragmatic eventration. Gastrointestinal signs: intestinal malrotation, umbilical hernia, omphalocele. Genitorenal disorders: bicornuate uterus, proximal non-acidotic tubulopathy. CNS: partial or complete agenesis of the corpus callosum, empty sella turcica, mental retardation. Laboratory data: proteinuria, urinary excretion of retinol-bound and vitamin D-bound proteins.
Molecular basis [31]: mutation in the low-density lipoprotein receptor-dependent protein 2 (LRP2) gene.
130720 Empty sella turcica primary syndrome. Lateral meningocele syndrome, Lehman syndrome. Clinically: defect of the dorsum sella, increased size of the sella turcica, internal auditory canal and optic nerve foramen, additional bones in the lambdoid suture, osteosclerosis, raised angles of the lower jaw, developmental anomalies of the spinal cord, cerebellum and cortex, multiple spina bifida, moderate growth retardation, facial hypoplasia, anti-Mongoloid eye section, gum hypoplasia, high arched palate, mandibular hypoplasia, enlarged foramen magnum, platybasia, basilar impression, enlargement of the spinal canal and intravertebral foramina, anomalies of the vertebral bodies [32–34].
We present a clinical case of PTS syndrome in a child.
Girl A., 10 years old, was admitted to the neurological department with complaints of myoclonic twitching of the shoulder girdle and arms, convulsive seizures with impaired consciousness. I was periodically bothered by headaches and decreased vision at dusk.
History of the disease. The child has been ill for a month, when single myoclonic convulsions appeared in the upper shoulder girdle. Then they became more frequent up to several times a day and became serial. Generalized convulsive seizures developed twice.
Anamnesis of life. Girl from first pregnancy, urgent birth. Birth weight - 3200 g. The umbilical cord is entwined around the neck. The Apgar score is 8 points. She did not lag behind in development.
Neurological status: short neck. Unusual shape and reduction in the size of the ears. Hypertelorism. Palpebral fissures S > D, pupils D = S. Posterior internuclear ophthalmoparesis. The left nasolabial fold is smoothed. Tendon reflexes are uniformly animated. Statics and coordination are not impaired. Hypermobile joint syndrome. Poor posture, lumbar hyperlordosis. Flat feet.
Electrocardioscopy: semi-vertical electrical position of the heart.
Electroencephalography: upon awakening and hyperventilation - short discharges of generalized bilateral synchronous peak-wave complexes without visible clinical manifestations. Photosensitivity was not detected. The pattern corresponds to one of the forms of idiopathic generalized epilepsy.
Ophthalmologist examination: VOU = 0.9. The fundus is normal. Slight narrowing of bitemporal visual fields.
Visual evoked potentials to the presentation of a chromatic and achromatic reverse checkerboard pattern (RP) showed interhemispheric asymmetry and prolongation of the latency of N75, P100, N145 (Tables 1–3).
Changes in latency rates were also detected upon presentation of red-blue and red-green chromatic PN, which confirms a visual impairment for colors (Tables 2–3).
A study of reverse PN by visual quadrants showed an extension of P100 latency from the temporal and inferior nasal quadrants of the left eye and the inferior temporal quadrant of the right eye (Fig. 1).
Visual cognitive evoked potentials (CEP): prolongation of P300, N400 latency during the study of abstract-verbal pattern, decrease in amplitude. Prolongation of sensorimotor reaction time (Table 4).
Auditory EPs correspond to age standards.
Psychological testing. MMSE: 26 points; FAB: 13 points; clock drawing test: 8 points; memorization test 10 words: maximum 7 words, unstable attention, extra words; Schulte tables: work efficiency 50 s - 4 points, mental stability - 1 point.
Conclusions: Mild cognitive impairment. Frontal dysfunction with impairment of semantic memory, conceptualization, verbal fluency, dynamic praxis. Decreased concentration. Lack of mental stability.
Ultrasound Dopplerography of the vessels of the neck and brain: cerebral arterial blood flow without pathology. Blood flow in the extracranial segments of the ICA is moderately accelerated, S > D. Signs of dystonia of the cerebral arteries and jugular veins. Obstruction of venous outflow from the anterior parts of the skull, S > D.
X-ray of the skull: the sella turcica is deep, the contours are clear, the dorsum is porous. The sella turcica index is 0.85 (normal is 0.84–1.0).
MRI of the brain: the pituitary gland is reduced in volume, homogeneous, spread across the bottom of the sella turcica. No additional formations or foci of pathological MR signal are detected in it. The suprasellar cisterns fill the cavity of the sella turcica. The bodies of the lateral ventricles are dilated to 1.3 cm (Fig. 2).
MRA: S-shaped tortuosity of the left ICA (Fig. 3).
In the blood: bilirubin - 9.8 µmol/l (indirect - 9.8, direct - 0), ALT - 33 U/l, AST - 16 U/l, blood glucose - 4.5 mmol/l.
Blood prolactin - 129.5 µIU/ml (normal range from 127 to 637 µIU/ml), FSH - 4.12 mIU/ml (normal range from 0.2 to 3.8 mIU/ml), luteinizing hormone - 0.138 mIU/ml (normal range from 0 to 0.2 mIU/ml), thyroid-stimulating hormone - 2.46 µIU/ml (normal range from 0.28 to 4.30 µIU/ml).
An examination by an endocrinologist did not reveal any pathology.
Therapy was carried out: convulex retard, diacarb, asparkam. The attacks stopped.
Thus, taking into account craniofacial and auricular stigmas, anomaly of color perception, cognitive impairment, epileptic seizures, the presence of primary PTS syndrome, we hypothesized a defect in VSX1, mapped to 20p11.2: (605020 MIM) Craniofacial anomalies including empty sella turcica, corneal endothelial changes and retinal and auditory bipolar cell abnormalities. However, the inability to detect the gene in our conditions led to the following formulation of the diagnosis: symptomatic epilepsy with frequent myoclonic and generalized convulsive seizures. Congenital empty sella syndrome. Congenital connective tissue dysplasia with hypermobile articular syndrome, tortuosity of the left internal carotid artery.
Emerging syndrome - what to do?
What is a Turkish nest and why is it special? Why is its emptiness during research alarming and even frightening?
This is a small depression in the sphenoid bone located at the temporal part of the skull. It is a small bony pocket located under the hypothalamus. It has a specific shape, from which the name comes.
On both sides of it pass the optic nerves, which cross over the area of the recess. In a normal state it is filled with the pituitary gland , in an abnormal state - with cerebrospinal fluid .
Let's understand the terminology:
- The pituitary gland is an endocrine gland. She is responsible for the production of hormones, metabolism and reproductive functions.
- The hypothalamus is a small region in the diencephalon. Controls the release of pituitary hormones. They are connected to each other by a leg. It is a link between the nervous and endocrine systems. Regulates the functions of hunger, thirst, sexual desire, sleep, wakefulness.
- The optic nerves have special sensitivity. Through them, visual stimulation is transmitted to the brain.
- Hormones are biologically active substances. They are produced by endocrine glands. Affect the physiological functions and metabolism of the body.
- The diaphragm is the dura mater. It covers the sella turcica and has a hole in the center through which the hypothalamus connects to the pituitary gland. Its attachment, thickness, and opening pattern have anatomical variations for each patient. And the absence or underdevelopment determines this type of syndrome.
- CSF is cerebrospinal fluid. When it gets into the sella turcica, the gland located there becomes deformed and decreases in vertical dimensions.
When the diaphragm subsides, due to its underdevelopment, the pituitary gland is pressed to the bottom. dysfunction develops . A detailed examination is carried out when the clinical picture is pronounced. Signs of the syndrome are caused by difficulty in the flow of hormones into the pituitary gland from the hypothalamus.
Incidence (per 100,000 people)
| Men | Women | |||||||||||||
| Age, years | 0-1 | 1-3 | 3-14 | 14-25 | 25-40 | 40-60 | 60 + | 0-1 | 1-3 | 3-14 | 14-25 | 25-40 | 40-60 | 60 + |
| Number of sick people | 3 | 5 | 7 | 13 | 15 | 18 | 21 | 6 | 10 | 14 | 16 | 26 | 36 | 42 |
Treatment
Therapy methods should be aimed at both eliminating the root cause of the disease and correcting neurological, ophthalmological and endocrine complications. The secondary form of pathology may be an indication for surgical intervention.
It should also be borne in mind that treatment is not required in all cases. If the patient does not exhibit symptoms and complications of sella turcica syndrome, then there is no need for therapy.
Drugs
Drug treatment can be aimed at eliminating the symptoms of the disease and correcting complications. Patients are prescribed hormonal drugs to improve endocrine status and non-steroidal anti-inflammatory drugs to relieve headaches. The treatment regimen depends on the complaints of the individual patient and the severity of the condition.
Surgery
Surgery is required for a complex form of the disorder, accompanied by the discharge of cerebrospinal fluid from the nose and other neurological lesions. During neurosurgical treatment, the natural anatomy of the sella turcica is restored.
Symptoms
The sella turcica contains the main center of the endocrine system, and next to it the optic nerves. In research, specialists are not interested in the structure of the recess, but in the diaphragm, the pituitary gland, its stalk, the hypothalamus and the specifics of the optic nerve.
When anomalies occur:
- Headache . First mild and intermittent, then constant and more severe. Dizziness and unsteadiness of gait appear.
- Vision problems manifest themselves in pain in the periocular area. There may also be loss of visual acuity, double vision, lacrimation, and the presence of a blurred vision. When examined by a specialist, a narrowing of the visual field and inflammatory processes in the optic nerve are detected.
- Endocrine disorders are caused by hormones. They can be produced both in deficiency and in excess. They lead to sexual dysfunction, menstrual irregularities, and diabetes insipidus.
If the pituitary gland is almost invisible on x-rays, then they say that the sella turcica is empty. The gland is spread out in the recess of the pocket in a thin layer. But clearly defined symptoms may not be observed. Just an ailment that can be attributed to anything, for example, overwork.
How dangerous it is for the patient
The risk of complications is higher if more than 90% of the pituitary gland is damaged due to its compression. Against the background of a stressful situation, patients may experience:
- severe vegetative crises leading to loss of ability to work;
- fainting conditions;
- personality changes;
- hypothyroid and thyrotoxic crises;
- sexual dysfunctions, infertility;
- progression of diabetes insipidus, Itsenko-Cushing's disease, acromegaly.
Visual impairment and the threat of blindness are indications for surgical treatment
Symptoms of SPTS
Usually the anomaly occurs unnoticed and does not cause any discomfort to the patient. Often, the syndrome is discovered by chance during a routine X-ray examination. The pathology occurs in 80 percent of women over the age of 35 who have given birth. Approximately 75% of them are obese. However, the clinical picture of the disease can vary dramatically.
Most often, the syndrome reduces a person’s visual acuity, causes a generalized narrowing of peripheral fields, and bitemporal hemianopsia. Patients also complain of frequent headaches and dizziness, lacrimation, and fogginess. In rare cases, swelling of the optic disc is observed.
Many nasal diseases result from rupture of the sella, which occurs due to excessive pulsation of the cerebrospinal fluid. In this case, the risk of developing meningitis increases several times.
Almost all endocrine disorders lead to pituitary dysfunction and the occurrence of SPTS. Among them are both rare genetic abnormalities and:
- increased, decreased levels of tropic hormones;
- excessive secretion of prolactin;
- metabolic syndrome;
- dysfunction of the anterior pituitary gland;
- increased production of adrenal hormones;
- diabetes insipidus.
The following disorders can be noticed from the nervous system:
- Regular headache. Occurs in approximately 39% of patients. Most often, it changes its location and strength - it can go from mild to unbearably regular.
- Disorders in the functioning of the autonomic system. Patients complain of surges in blood pressure, dizziness, spasms in various organs, and chills. They often lack air, feel unreasonable fear, and are overly anxious.