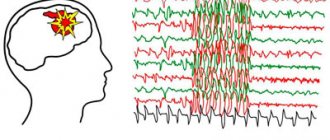
Juvenile myoclonic epilepsy is a form of generalized epilepsy, the basis of the clinical picture of which is myoclonic seizures - asynchronous muscle contractions that occur briefly in symmetrical areas of the body, mainly in the arms and shoulder girdle. Along with myoclonic episodes, absence seizures and clonic-tonic generalized epileptic seizures can be observed in the clinic. Juvenile myoclonic epilepsy is diagnosed based on the clinical picture of the disease and the results of electroencephalography, with the exclusion of organic cerebral pathology according to a neurological examination and MRI. Treatment is carried out mainly with valproic acid preparations. As a rule, lifelong monitoring by an epileptologist is necessary.
Case history and nomenclature
Benign myoclonic epilepsy syndrome in children (BMSES) was not clearly defined until it was first described in 7 young children in 1981 (Drave and Bior, 1981).
In this article, SDMED was defined as the occurrence of myoclonic seizures (MS) without seizures other than simple febrile seizures (FS) in the first three years of life in healthy children. These MPs were easily treatable and became quite rare during subsequent years of childhood. Psychomotor development remained normal, and no severe psychological consequences were observed. Since then, many other cases have been described in the literature. SDMED is included in the 1989 International Classification of Generalized Idiopathic Epilepsies (Commission, 1989). Some authors have described reflex MP in patients caused by noise or conversation, and proposed to distinguish between two separate nosological forms, and the 2nd was called reflex myoclonic epilepsy in children (Ricci et al., 1985). We think that such a division is inappropriate and will describe all cases as SDMED. To our knowledge, 98 cases have been described in the literature to date, of which 88 fit the classic description of SDMED and 10 were defined as “reflex SDMED.”
There are now 5 new patients under our supervision, four of whom have spontaneous and reflex seizures, which brings the total number of patients to 103.
The first description of SDMED indicated that the onset of the disease was before 3 years of age, while subsequent reports suggested a later onset in some patients, up to 4 years 8 months (Giovanardi-Rossi et al., 1997). This means that epilepsy of the same type can appear at different ages (Guerini et al., 1997).
General provisions
Epidemiology
According to the few epidemiological data available, SDMED accounts for less than 1% of all epilepsies (St. Paul's Center, 1997), 1.3% and 1.72% of epilepsies that begin in the first year of life (Dalla Bernardina et al., 1983). , and 0.39% of epilepsies beginning in the first 6 years of life (Ohtsuka et al., 1993).
Floor
Gender distribution: 52 boys and 27 girls.
Genetics
The genetics of SDMED are unknown. There are few patients, and cases of familial SDMED have not been described. A family history of epilepsy or febrile seizures (AF) was present in 39% of 80 patients. In 70 patients, the incidence of AF in the family was 17%, and the incidence of epilepsy was 24%. It is difficult to assess the type of epilepsy found in relatives. In 10 cases it was probably idiopathic epilepsy. In the case reported by Arzimanoglou et al (1996), the proband was the 2nd of 2 brothers, and the eldest suffered from typical epilepsy with myoclonic-astatic seizures (EMAS, Douz syndrome).
Anamnesis of life
Most patients had no history of pathology before the onset of MP. Only two (1.9%) had comorbidities: Douz syndrome (Drave et al., 1992) and hyperinsulin diabetes (Colamaria et al., 1987).
However, cases of AF are not rare: 19 out of 64 patients (30%). AFs were always simple, but rare (1-2) and were observed before the onset of myoclonus and before the start of treatment (15 cases).
Clinical and EEG manifestations
The age at which the disease begins is usually from 4 months to 3 years. Rarely does it start earlier. Later onset was reported by Giovanardi-Rossi et al (1997) - 4 years 9 months - and Lin et al (1998) - 3 years 9 months.
At first, MPs are short, most often sparse, involving the upper limbs and head, and rarely the lower limbs. In infants they are subtle and parents sometimes find it difficult to determine their onset and frequency. They often talk about "spasms" or "head bobbing." Later, the frequency of attacks increases.
Video-EEG recordings allowed us to conduct a precise analysis of these seizures. These are more or less massive myoclonic jerks affecting the trunk and limbs, causing tilting of the head and upward and outward movement of the upper limbs with flexion of the lower limbs, and sometimes rotation of the eyeballs. Their intensity is different for each child; in the same patient it can change with each attack. In severe forms, objects suddenly fall out of the hands, and sometimes the patient falls. In moderate forms, only a short forward movement of the head or even a simple closing of the eyes is noted; as a rule, the seizures are very short (1-3 sec.), although they can last longer (in older children) and consist of pseudorhythmic twitchings that do not last. more than 5-10 sec. They develop several times a day at different intervals. Unlike childhood spasms, they do not occur in long series and do not develop at the moment of awakening, but rather during sleep. In some patients, sudden noise, sudden contact, or repeated photostimulation (RPS) can trigger seizures. With single seizures, it is difficult to assess the state of consciousness. Only with repeated seizures is there a slight disturbance of consciousness without interruption of activity. We did not observe the sudden short vocalisms reported by Lin et al. (1998). The authors point to the involvement of the diaphragm and/or abdominal muscles in the pathological process, causing expiratory noise. In reflex MP, myoclonus can be induced during both wakefulness and sleep, the excitability threshold is low in stage 1, but it gradually increases during the slow stages (Ricci et al., 1995). In patients with reflex MP, the REM sleep phase was not recorded or analyzed. Because The child’s development is normal, parents and pediatricians mistake these movements for pathology.
The EEG picture outside the attack is within normal limits.
Myoclonus is always associated with an EEG discharge. Recordings show that myoclonus is accompanied by a discharge of fast generalized spike waves (SW) or polyspike waves (PSW) with a frequency of more than 3 Hz, with the same duration as myoclonus. This is a more or less regular discharge and can begin in the frontal areas and in the vertex. Myoclonus is short (1-3 seconds) and usually isolated. The myoclonic jerk may be followed by brief atonia. Sometimes after an attack, voluntary movements appear, regarded as normal muscle contractions. And in only one patient we noted a combination of myoclonus in the deltoid muscle with pure atony of the cervical muscles. During drowsiness, myoclonus increases, but not always. They disappear during slow-wave sleep. MPs caused by tactile and auditory stimuli have the same characteristics. Ricci et al. (1995) indicate that the primary manifestations are usually, but not always, manifested only by the blink symptom, followed by 40-80 ms. followed by a myoclonic jerking of the arm. After a myoclonic attack, a refractory period begins, lasting from 20-30 seconds. up to 1-2 minutes, during which external stimuli, even fear, do not provoke attacks. Repeated photostimulation (RP) can also provoke MP (Drave et al., 1992; Todt and Müller, 1992; Giovanardgi-Rossi et al., 1997; Lin et al., 1998).
Interictal EEG data without pathology. Spontaneous SV discharges are rare; Slow waves can be detected in the central areas of the brain. PF causes SV without existing concomitant myoclonus. Short nap recordings showed normal sleep organization; generalized SV discharges may appear during REM sleep.
Juvenile myoclonic epilepsy - symptoms and treatment
The main diagnostic criterion for the disease is the presence of myoclonic seizures.
History taking
At the appointment, the doctor asks about unusual sudden conditions:
- tremors in the body;
- déjà vu - a state in which a person feels that he has once been in a similar situation or place;
- loss of consciousness, etc.
Patients may not pay attention to such symptoms and consider them to be their peculiarity. Absence seizures and generalized tonic-clonic seizures with loss of consciousness, especially during sleep, they may completely forget. Therefore, when collecting anamnesis, it is important to find out the circumstances of the attack not only from the patients themselves, but also from relatives and eyewitnesses.
Electroencephalogram (EEG)
The main way to diagnose epilepsy is an electroencephalogram - a research method that records the total electrical activity of cells in the cerebral cortex.
Now the diagnosis of epilepsy is established using long-term video-EEG monitoring - an electroencephalogram is recorded in parallel with one or more video cameras, an ECG sensor and, if necessary, additional monitoring of muscle activity, frequency and depth of breathing.
The main background of bioelectrical activity in juvenile myoclonic epilepsy, as a rule, corresponds to the age norm. Pathological activity is manifested by short and generalized discharges of polyspikes (sharp-wave complexes), which are recorded during myoclonic jerks and polyspike-wave complexes between attacks.
The phenomenon of photosensitivity often occurs in the disease. To detect it during an EEG, the patient is asked to close his eyes and undergo rhythmic photostimulation with a frequency of about 15 Hz [16].
Epileptic photosensitivity is a predisposition to seizures when exposed to light. It can be asymptomatic or manifest as epileptic seizures under the influence of provoking factors: video games, working on a computer, watching TV, flashing lights in nightclubs and natural light.
MRI does not reveal pathological changes in the brain in juvenile myoclonic epilepsy [17].
The intelligence and neurological status of the disease are normal. Expressed emotional instability and signs of neurotic personality development: sudden changes in mood, short temper and increased anxiety
Course and treatment
Children with SDMED do not experience seizures of any other type, even if they remain untreated (up to 8.5 years in one of our patients), this is especially true for petit epileptic or tonic seizures. Clinical examination results are normal. Interictal myoclonus was described only by Giovanardi-Rossi et al. (1997) in 6 patients. Analyzing the condition of our patients, we found mild interictal myoclonus in 2 according to EEG recordings. Many patients were not examined, but when CT and MRI were performed, the results were normal (33 patients).
Outcome likely depends on early diagnosis and treatment. Myoclonus is easily amenable to monotherapy with valproate, and the child’s development is appropriate for his age. If left untreated, the patient continues to have myoclonic seizures, which can lead to impaired psychomotor development and behavioral abnormalities.
Treatment methods were more or less thoroughly tested on 74 patients. 65 patients received monotherapy, 6 received polytherapy, and 3 received no treatment. Monotherapy included valproate (VPA), phenobarbital (PB), nitrazepam (NTZ). 6 patients received therapy with primidone (PRM) and ethosuximide (ESM). As a result of treatment, seizures disappeared in 69 patients (93%).
These data confirm that VPA is the first choice drug for SDMED. However, treatment should be carried out under the control of drug concentration in plasma, because improper use may lead to relapse or cause drug-resistant epilepsy.
Long-term results and prognosis
The duration of observation for 63 patients was 9 months. up to 27 years, of which 45 patients were observed for about 5 years. The age of patients during the observation period ranged from under 5 to over 15 years.
In all cases, MPs were stopped. The duration of the disease is known in 52 patients: in most of them, MP lasted less than one year, in 7 - from 1 to 2 years, only in 5 - more than two years.
The occurrence of generalized epileptic seizures (GES) after cessation of MP was reported in the case of 10 patients without concomitant MP.
The results of observation of 74 patients were reported.
The attacks stopped in 28 patients over the age of 6 years. In 6 children of the same age, seizures persist: with photostimulation - in 2 (Drave et al., 1992), for an unknown reason - in 3 (Giovanardi-Rossi et al., 1997). Also, attacks persisted in 13 patients under 6 years of age. 3 patients under 6 years of age remained without treatment (Ricci et al., 1995). There is no information about 24 patients.
The EEG pattern is known for 55 patients. It quickly normalized in 23. Rare spontaneous generalized SV persisted in 13. Photosensitive changes were observed in 6. Interestingly, photosensitivity can appear after the disappearance of MP and persist for many years after the cessation of MP until adulthood. Focal abnormalities were also presented. They were recorded in 5 patients during awakening as SV in two fronto-central and parietal areas, sometimes in the fronto-parietal and fronto-temporal. They disappeared over time. On the contrary, in other patients they appeared only during sleep. They still persisted at the end of the observation period in 5 of our patients, and only during sleep.
Overall, the psychological outcome was favorable and most patients recovered. This is known for sure in 69 cases. 57 (83%) were healthy, of whom 38 were aged 5 years or older. 10 (14%) had mild retardation and attended a specialist school, but none of them were admitted to hospital. In 2 patients (3%) cognitive ability was impaired and deviations in personality formation were identified: one patient had Down syndrome and severe sensitivity, the other had MP with sensitivity to IRS and to eye closure up to 5 years of age. This psychotic disorder appeared in him at the age of 10 years and progressed.
This psychological outcome depends in part on early diagnosis, allowing appropriate treatment and reassuring the family of a good future.
These data confirm the generally good prognosis of SDMED. Seizures caused by noise or conversation were easier to control than spontaneous ones. Photosensitivity was more difficult to control and persisted for several years after the seizures stopped.
Epilepsy was known back in ancient Babylon (the so-called “falling” disease), and Hippocrates described it as a disease of the brain. The notorious John Hughlings Jackson more than 100 years ago described epilepsy as “periodically occurring excessive and disorderly discharges of nervous tissue.” Nowadays, epilepsy refers to conditions characterized by repeated relatively stereotypical seizures [1].
Epilepsy (or rather, epilepsy) is a group of diseases in which the main manifestations are repeated paroxysmal seizures. The prevalence (occurrence) of epilepsy among children is undoubtedly high, but accurate statistics depend on whether the so-called cases are included in the established diagnoses. single (isolated) seizures or non-convulsive seizures, as well as febrile seizures. For example, according to data from Millichap JG (1968), febrile seizures account for up to 2% of all diseases in childhood [2]. In any case, epilepsy is the most common of the serious paroxysmal disorders of brain function, with a prevalence in the general population of 0.3% to 2%, and in children about 1% [3,4,5].
Etiology and pathogenesis
In the occurrence of epileptic paroxysms in children, as in adults, the main importance is given to hereditary or acquired predisposition, in addition, various exogenous influences (craniocerebral injuries, viral and bacterial infections, etc.) play a certain role.
Epileptic activity itself is caused by metabolic disorders in synapses and mitochondria, leading to the so-called. “epileptization” of neurons in various (most predisposed to this) brain structures through both synoptic and non-synaptic activation with the involvement of new formations of the central nervous system in the pathological process [3,4].
Classification of epilepsy
Currently, epilepsy in children is divided in accordance with the classification adopted in 1981 in Kyoto (Japan). Among them are: I. Partial (focal, local) seizures: A) simple partial; B) complex partials; C) Partial with secondary generalization; II. Generalized seizures: A) absences (typical and atypical); B) myoclonic seizures; C) clonic seizures; D) tonic seizures; E) tonic-clonic seizures; F) atonic seizures; III. Unclassified epileptic seizures. Menkes JH and Sankar R. propose to distinguish between primary (idiopathic), secondary (symptomatic) and reactive epilepsy. All of the listed epilepsies may be characterized by generalized or partial (focal) seizures [1].
The classification of epilepsies, epileptic syndromes and similar diseases adopted by the International League Against Epilepsy (1989, New Delhi, India) provides for the following headings: 1) localization-related forms (focal, focal, local, partial): 1.1. idiopathic (with age-dependent onset) - benign childhood epilepsy with central temporal peaks (Rolandic), childhood epilepsy with occipital paroxysms, primary reading epilepsy; 1.2. symptomatic - chronic progressive partial epilepsy of Kozhevnikov, seizures characterized by specific methods of provocation, other forms of epilepsy with a known etiology or organic changes in the brain (frontal, temporal, parietal, occipital); 1.3. cryptogenic; 2) generalized forms of epilepsy: 2.1. idiopathic (with age-dependent onset) - benign familial convulsions of newborns, benign convulsions of newborns, benign myoclonic epilepsy of infancy, childhood absence epilepsy (pycnolepsy), juvenile absence epilepsy, juvenile myoclonic epilepsy, epilepsy with generalized convulsive seizures of awakening, other idiopathic generalized forms of epilepsy, forms not mentioned above, characterized by specific methods of provocation (more often - photosensitivity epilepsy); 2.2 cryptogenic and/or symptomatic - West syndrome (infantile spasms), Lennox-Gastaut syndrome, epilepsy with myoclonic-astatic seizures, epilepsy with myoclonic absence seizures; 2.3. symptomatic: 2.3.1. nonspecific etiology - early myoclonic encephalopathy, early infantile epileptic encephalopathy with a burst-suppression pattern on the EEG (Otahara syndrome), other symptomatic generalized forms of epilepsy not mentioned above; 2.3.2. specific syndromes; 3) epilepsy that does not have a clear classification as partial or generalized: 3.1. having both generalized and partial manifestations - neonatal seizures, severe myoclonic epilepsy of early childhood, epilepsy with continuous peak waves during slow-wave sleep, acquired epileptic aphasia (Landau-Kleffner syndrome), other unclassified forms of epilepsy not defined above; 3.2. attacks that do not have clear generalized or partial signs; 4) specific syndromes: 4.1. situationally determined seizures - febrile convulsions, seizures that occur only as a result of acute metabolic or toxic disorders; 4.2. isolated seizures or isolated status epilepticus.
Clinical picture
Due to the large number and variety of forms of epilepsy known today, we will dwell only on some of the features of epileptic seizures in children. Let us note that in children of the first year of life, due to the predominant functioning of the brain stem structures, tonic seizures are usually observed, and the clonic component of the seizure is formed at an older age.
With generalized seizures, the child usually suddenly loses consciousness and falls, groaning or screaming. The tonic phase of the seizure begins, when a sharp tension of the entire muscles is noted: throwing the head back, cyanosis of the face with a grimace of fear, dilation of the pupils with a lack of reaction to light, clenching of the jaw, bending of the arms at the elbow joints, clenching of fists, stretching of the lower extremities. This is followed by the clonic phase: convulsions of the upper and lower extremities, head, sometimes involuntary urination and defecation occur. Gradually, cramps are curtailed with a decrease in severity (muscles relax, tendon reflexes decrease). The child stops responding to the environment and falls asleep (or regains consciousness in a state of complete amnesia).
Absences (minor seizures) are manifested by a short-term (several seconds) loss of consciousness followed by amnesia. In this case, convulsions and other motor disorders may be absent, although motor phenomena of varying duration, individual myoclonic convulsions, elementary automatisms, vegetative-visceral or vasomotor disorders are often observed.
Myoclonic spasms (infantile spasms) are characterized by the presence of peculiar spasms (mainly of the flexion type), a progressive course, as well as specific changes in the EEG (hypsarrhythmia).
The manifestations of partial seizures in children depend on the preservation or loss of consciousness during the seizure (simple partial without impairment of consciousness or complex partial in the form of Jacksonian, adversive, etc. with impairment of consciousness).
Diagnostics
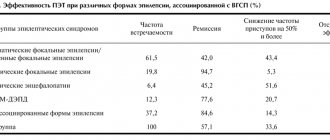
Modern methods for diagnosing epilepsy in children and adolescents involve the widespread use of methods such as electroencephalography (EEG), computed tomography (CT), and magnetic resonance imaging (MRI). If necessary, EEG video monitoring, positron emission tomography (PET), as well as a number of biochemical immunological and other special research methods are used.
Treatment of epilepsy in children
General principles of treatment of epilepsy include: 1) adherence to a general and nutritional regimen, 2) therapy with anticonvulsants and additional individual drug treatment, 3) psychotherapy.
The principles for prescribing anticonvulsants to children with epilepsy are as follows [1]:
- The choice of drug is determined by the type of seizure and the potential toxicity of the drug.
- Treatment should begin with one drug, increasing its dosage until the attacks stop (or the toxic effect of the drug appears). If the prescribed drug does not control the attacks, it is gradually withdrawn (as another drug is prescribed and its dose is increased).
The concept of monotherapy for epilepsy (ie the predominant use of one anticonvulsant drug to treat seizures) has received widespread support worldwide. It is supported by the results of numerous clinical studies that document various “errors” of polytherapy. First, chronic drug toxicity is directly related to the number of drugs consumed by the child. Even if none of these drugs is prescribed at a dose that could cause toxicity, their effects on sensory and intellectual functions appear to be cumulative. Secondly, polytherapy can lead to aggravation of attacks in a significant proportion of patients (especially children). Third, with polytherapy it can be difficult (or impossible) to identify the cause of adverse reactions.Reynolds EH and Shovron SD (1981) indicate that the transition from polytherapy to monotherapy is accompanied by better control of seizures and improvement in indicators of intellectual development (cognitive functions) [6]. Hanson RA and Menkes JH 30 years ago conducted a comparative analysis of the effectiveness of mono- and polytherapy. It turned out that in only a quarter of cases, one anticonvulsant was not enough to control seizures and a second drug was required [7].
- Changes in the dose of the drug(s) should be made gradually, usually no more than once every 5-7 days.
- It is unlikely that seizure control will be achieved with a lesser known drug if the first choice drug is not suitable. At the same time, the likelihood of unusual adverse reactions when prescribing a new or rarely used drug is very high.
- Having achieved control over the attacks, you should adhere to the use of the prescribed drug for a long time.
- In patients taking phenobarbital, phenytoin, carbamazepine, and ethosuximide, blood levels of anticonvulsants are extremely important. These concentrations should be monitored frequently, especially in patients not responding to therapy or showing signs of drug toxicity.
- It is known that during treatment with anticonvulsants, disorders of the blood and liver function often develop. They are preceded by the so-called. asymptomatic phase, which can be identified by laboratory and instrumental methods. In this regard, routine blood tests and liver assessment are suggested. At the same time, some American researchers indicate that if every patient receiving anticonvulsants is examined in full, then the cost of epilepsy therapy will become truly astronomical [8].
- Anticonvulsants should be discontinued with caution and gradually. Abrupt withdrawal of the drug, especially when it comes to barbiturates, is the most common cause of the development of status epilepticus [1].
Below, in alphabetical order, is information about the main drugs used in the treatment of epileptic syndromes in children.
Adrenocorticotropic hormone (ACTH, corticotropin, adrenocorticotropin, ACTH-gel, synacthen-depot)
Indications: 1st choice drug in the treatment of infantile spasms (West syndrome) and other infantile epileptic encephalopathies, 2nd choice drug in the treatment of Lennox-Gastaut syndrome, childhood form of Kozhevnikovsky epilepsy, epileptic aphasia (Landau-Kleffner syndrome), myoclonic-astatic epilepsy.
Dosage: within 1 week of treatment - 150 U/sq.m, 2 - 75 U/sq.m, 3-4 - 75 U/sq.m, 5-6 - 50 U/sq.m, 7-8 - 40 U/sq.m, 9-10 - 20 U/sq.m, 11-12 - 10 U/sq.m of body surface every other day.
Acetazolamide
Indications: for large, small and partial complex seizures, for catamnial seizures as a drug of 2 choice.
Dosage: 10-15 mg/kg/day (in 1-3 doses).
Note: Acetazolamide should be taken with potassium supplements.
Benzobarbital
Indications: for generalized tonic-clonic, myoclonic, simple partial and psychomotor seizures.
Dosage: (3-6 years) 0.1-0.15 g/day, (7-10 years) 0.3-0.5 g/day, (11-14 years) 0.3-0.4 g /day (take 2-3 times a day).
Bromides/bromine salts (sodium bromide, potassium bromide)
Indications: as a drug of 3 choice in the treatment of large, less often focal seizures.
Dosage: 75 mg/kg/day (in 1-2 doses).
Note: Bromides are the first choice drugs for the treatment of intermittent porphyria.
Valproic acid (Depakine, Depakine-Chrono, Depakine-Enteric, Apilepsin, Acediprole, Convulex, Convulsofin, Orfiril, Encorate)
Indications: pycnoleptic absence seizures, myoclonic-astatic, generalized photosensitivity, myoclonic, grand tonic-clonic seizures, infantile spasms, simple and complex partial seizures. Dosage: 30-40 mg/kg/day Depakin-chrono - 1-2 times a day. Other drugs - 2-3 times a day.
Note: if necessary, the dose can be increased.
Vigabatrin
Indications: partial and secondary generalized paroxysms resistant to other anticonvulsants, West syndrome, Lennox-Gastaut syndrome.
Dosage: from 40 to 100 mg/kg/day (in 1-2 doses).
Note: the drug is ineffective for myoclonic seizures and absence seizures. More often used as part of polytherapy.
Gabapentin
Indications: (for children over 12 years of age) resistant (to other anticonvulsants) forms of partial and secondary generalized epileptic seizures, Lennox-Gastaut syndrome (as polytherapy).
Dosage: 10-30 mg/kg/day.
Note: The drug is not registered in the Russian Federation.
Diazepam (seduxen, relanium, sibazon, apaurin, valium, diazepex, diapam, dicam, calmpoose)
Indications: for all forms of status epilepticus (iv and per rectum), as well as per os for a short time in the treatment of febrile convulsions, increased frequency of seizures (1-2 times a day).
Dosage: for the treatment of status epilepticus intravenously or rectally - children 1 year of age - 0.4 mg/kg/day, children over 1 year - 0.3 mg/kg/day. As part of polytherapy, at the age of 1-3 years - 2-3 mg/day, 3-7 years - 5-7 mg/day, 7 years and older - 8-16 mg/day.
Carbamazepine
Indications: generalized tonic-clonic, simple partial, complex, secondary generalized, psychomotor seizures.
Dosage: 30 mg/kg/day (in 1-3 doses).
Clonazepam
Indications: for all forms of status epilepticus (iv and per rectum), as an additional drug in the treatment of generalized epilepsy with myoclonic-astatic seizures, for myoclonic, simple and complex partial seizures per os (in 1-2 doses).
Dosage: for the treatment of status epilepticus 0.25-0.5 mg in 30 seconds, for oral administration - 0.15 mg/kg/day.
Lamotrigine
Indications: treatment of partial, complex partial, secondary generalized and primary generalized seizures, atonic seizures, absences resistant to therapy with other anticonvulsants, as well as Lennox-Gastaut syndrome (children over 2 years old).
Dosage: during therapy with lamotrigine and sodium valproate in combination with or without other antiepileptic drugs: 1-2 weeks - 0.15 mg/kg/day, 3-4 weeks - 0.3 mg/kg/day (given in 1 dose ), to achieve a therapeutic effect, the dose is increased by 0.3 mg/kg/day every 1-2 weeks to a maintenance dose of 1-5 mg/kg/day, but not more than 200 mg/day (in 1-2 doses); during therapy with lamotrigine and antiepileptic drugs that induce liver enzymes, in combination with or without other antiepileptic drugs (except sodium valproate): 1-2 weeks - 0.6 mg/kg/day, 3-4 weeks - 1.2 mg /kg/day (in 2 doses), to achieve a therapeutic effect, the dose is increased by 1.2 mg/kg/day every 1-2 weeks to a maintenance dose of 5-15 mg/kg/day (in 2 doses), but not more than 400 mg/day.
Notes: used when conducting polytherapy when first-choice drugs are ineffective (children over 2 years old).
Midazolam
Indications: in the treatment of status epilepticus (all forms), as an additional drug for all forms of seizures (especially myoclonic seizures).
Dosage: when prescribed per os, per rectum - (up to 12 years of age) - 7-15 mg/day, (after 12 years of age) - 15-45 mg/day.
Note: for the treatment of status epilepticus in children - 0.15-0.3 mg/kg (intramuscular or rectal).
Nitrazepam (radedorm, nitrazepam, eunoctin, berlidorm, nitrazadone, nitram, nitrosan)
Indications: as an additional remedy for (polytherapy) generalized epilepsy with myoclonic-astatic seizures, for myoclonic, simple and complex partial seizures.
Dosage: 0.4-0.5 mg/kg/day (in 1-2 doses).
Primidone (hexamidine)
Indications: in the treatment of generalized tonic-clonic, myoclonic, psychomotor and simple partial seizures.
Dosage: 20 mg/kg/day (in 1-2 doses).
Tiagabine (gabitril)
Indications: simple partial, complex, secondary generalized, psychomotor epileptic seizures.
Dosage: 0.5-1.0 mg/kg/day.
Note: the drug is not registered in the Russian Federation.
Topiramate
Indications: (children over 12 years of age) in the treatment of simple and complex seizures (with and without generalization), major tonic-clonic seizures, astatic seizures in Lennox-Gastaut syndrome, as an additional drug in the treatment of resistant epileptic syndromes.
Dosage: 200-400 mg/day (in 2 doses).
Phenytoin (diphenin)
Indications: for the treatment of generalized tonic-clonic, simple and complex partial, as well as psychomotor seizures.
Dosage: 7 mg/kg/day (in 1-2 doses).
Note: It is not recommended to administer in the same dose with other drugs (to avoid precipitation).
Phenobarbital (luminal, Sireysky mixture, gluferal, pagluferal 1, pagluferal 2, pagluferal 3)
Indications: in the treatment of generalized tonic-clonic epileptic seizures, simple partial and psychomotor seizures.
Dosage: newborns and infants - 10-15 mg/kg/day, children over 1 year - 3-5 mg/kg/day (in 1-2 doses).
Note: in young children the drug is characterized by pronounced rachitogenic activity.
Ethosuximide
Indications: in the treatment of pycnoleptic absences, myoclonic-astatic and myoclonic seizures.
Dosage: 30 mg/kg/day (in 1-2 doses).
Note: the initial dose of ethosuximide is 10-15 mg/kg/day during the first week of treatment, followed by increasing the dose to the therapeutic dose [3,9,10].
Non-drug treatment of epileptic syndromes comes down to prescribing a ketogenic diet or, in some cases, neurosurgical operations.
Despite the emergence of new, highly effective anticonvulsants, the ketogenic diet, based on reproducing the states of ketosis and acidosis during fasting, remains a very attractive alternative to drug treatment in the treatment of Lennox-Gastaut syndrome and other refractory epileptic syndromes. The mechanism of seizure control with this dietary therapy remains unclear. However, in the 90s. last century, the popularity of ketogenic diets in a number of countries has increased significantly [11]. This diet is considered particularly effective in the treatment of children aged 2-5 years with small motor seizures (it is difficult to maintain a state of ketosis in older children) [1]. Unfortunately, ketogenic diets have not yet found application in pediatric neurology in Russia.
Neurosurgical methods for treating epilepsy are not widely used in our country. However, there is evidence that hemispherectomy is effective for Rasmussen syndrome (chronic focal encephalitis) [1]. Modern world literature also reports the use of the following types of neurosurgical operations for various treatment-resistant epileptic syndromes: anterior temporal lobectomy [12], limited temporal resection [13] and extratemporal neocortical resection [14]. Stimulation of the vagus nerve occupies a special place among surgical treatment methods [15].
Differential diagnosis
If myoclonus begins in the first year of life, it must be differentiated from cryptogenic infantile seizures (CS). DS are clinically different from benign myoclonus: they are more severe and include tonic convulsions throughout the body, which is never observed in SDMED; single seizures are always combined with serial seizures in a given child; the occurrence of long serial spasms occurs after waking up. SDs then show the typical pattern of short tonic contractions on EEG, which is well described by Rusco and Vigevano (1993); Prolonged myoclonus rarely occurs. Ictal EEG variable: sudden intermittent hypsarrhythmias leveled off by superimposed fast rhythms, a large slow wave followed by leveling off or no visible change. The onset of DS is associated with behavioral changes, difficulty in speech contact, and a slowdown in psychomotor achievements.
The interictal EEG always has pathological signs: true hypsarrhythmia, or modified hypsarrhythmia, or focal disturbances; they do not detect individual or short bursts of bilateral synchronous SV or SDMED.
If psychomotor development and EEG are within normal limits on several examinations performed during wakefulness and sleep, seizures resembling DS should suggest the diagnosis of benign nonepileptic myoclonus, as described by Lombroso and Feuerman (1977). These patients even have ictal EEG without pathological changes (Drave et al., 1986; Pashats et al., 1999).
In the first year of life, myoclonic epilepsy of infancy may develop; it always begins with prolonged and frequent febrile seizures, and not with individual myoclonic seizures. In the second year, psychomotor development slows down.
If myoclonus begins after the first year of life, cryptogenic Lennox-Gastaut syndrome (CLG) may be suspected. In LGS (Byman and Dravet, 1992), the seizures are mainly not myoclonic, but myoclonic-atonic, and more often tonic, leading to sudden falls and injuries. Their EEG pattern is heterogeneous, and in the ictal period the high-amplitude either levels off, or high slow-wave activity is determined, followed by low-amplitude waves. At the very beginning, interictal EEGs may be normal, but later typical diffuse discharges of slow CO may appear. Typical electroclinical signs during sleep may be delayed in time. The diagnosis is based on the rapid association of seizures of different types, such as atypical absence seizures and axial tonic seizures, persistent behavioral disturbances and the emergence of new skills and the ineffectiveness of antiepileptic drugs.
If myoclonic seizures are isolated or associated with PEP, a diagnosis of myoclonic astatic epilepsy of early childhood (EMAI) should be considered, although in this syndrome myoclonic astanic seizures rarely begin before age 3 years (Douze, 1992). There are two significant differences: 1) the clinical aspect of seizures with stupor, which is not observed in SDMED (Guerrini et al., 1994); 2) EEG signs are also different: SV and PSV are more numerous, grouped into long flashes, connected by a typical theta rhythm over the centro-parietal zones. But some patients should probably be classified as SDMED. Similarly, Delgado-Escueta et al. (1990) included in the study or patient group, probably under the name of myoclonic epilepsy of childhood (MECD), cases of both EMAP and SDMED.
Finally, other epilepsies that begin in the first three years of life, in which myoclonus is the main seizure type and which have a variable prognosis, should be considered. They are heterogeneous: a combination of seizures of other types, persistent focal abnormalities on the EEG, delayed psychomotor development in the past, poor response to treatment, unclear prognosis (Drave et al., 1992).
Myoclonic-astatic seizures
Generalized epilepsy with myoclonic-astatic seizures: diagnosis and therapy
K.Yu. Mukhin, A.S. Petrukhin, E.A. Rykova
Myoclonic seizures (MS) are a common manifestation of many forms of childhood epilepsy (Guerrini). In this regard, an important task is to systematize various epileptic syndromes manifested by ioclonic seizures. Various approaches have been taken to systematize these types of seizures. It was proposed to distinguish symptomatic, cryptogenic idiopathic, as well as primary and secondary generalized forms of myoclonic. In recent years, with the introduction of video-EEG monitoring and intravital brain imaging methods (CT, MRI, PET) into clinical practice, it has become possible to more accurately differentiate various epileptic syndromes. The modern classification of epilepsy (1989) identifies the following epileptic syndromes in children, manifested by MP:
early myoclonic encephalopathy;
benign and malignant (severe) myoclonic epilepsy of infancy;
epilepsy with myoclonic absence seizures;
Lennox-Gastaut syndrome (LGS);
juvenile myoclonic epilepsy;
epilepsy with myoclonic-astatic seizures (MAE).
The first report of MAE was made by Jackson in 1886. The author observed a 7-year-old child, “who had been experiencing attacks of falls since he was 2.5 years old.” It was only in 1964 that Doose presented a detailed description of the syndrome and identified MAE under the name “akinetic myoclonic petit mal.” According to the author's original description, this form of epilepsy is characterized by a high genetic predisposition, the onset of attacks in preschool age; manifests itself predominantly in myoclonic and myoclonic-astatic seizures and has an unfavorable prognosis. At the same time, the issue of nosological independence of MAE is still being debated, which is not recognized by all authors. There are no clear diagnostic criteria for this form of epilepsy and, in particular, how it differs from the so-called “myoclonic variant” of LGS. The purpose of this study was clinical systematization, study of electroneurophysiological and neuroradiological features of MAE, as well as therapeutic tactics.
We observed 11 patients with MAE aged 4-15 years (average - 7.4+/-1.5 years), of which 7 were male and 4 were female. Video monitoring of seizures, EEG study (Medicor, 18 channels), EEG mapping and computed tomography of the brain (SOMATOM with a scanning step of 4 and 8 mm) were carried out. The results obtained were processed on an IBM PC 386 using the PARADOX database and the QUATRO-PRO statistical package.
Research results
Among 196 children with generalized forms of epilepsy examined at the epileptic center, MAE was detected in 11 cases, which is 5.6%. According to the anamnesis, pathology of the perinatal period was observed in the majority of patients (54.5%). Disorders during pregnancy dominated: toxicosis, threat of miscarriage (with the use of hormonal drugs). In 18.2% of cases, manifestations of birth trauma were noted. No familial cases of epilepsy were identified. The onset of epilepsy varied from 11 months. up to 5 years, averaging 2.3+/-1.90 years. In 81.8% of cases, the onset of the disease occurred in the age interval of 1-3 years. In the vast majority of cases (81.8%), MAE began with generalized convulsive seizures (GSE). In one patient the disease began with absence seizures and in one with tonic-clonic febrile paroxysms. The average age of onset of GSP was 2.4+/-0.38 years, absence seizures - 4.0+/-0.57 and myoclonic seizures -4.0+/-0.40. Clinical manifestations of MAE were polymorphic and included different types of seizures: myoclonic, myoclonic-astatic, typical absences, GSP and partial paroxysms. The core of MAE were myoclonic and myoclonic-astatic seizures. However, in no case did the disease begin with these attacks. MPs were characterized by short, lightning-fast, usually asynchronous and arrhythmic twitching of small amplitude in the legs and arms (hands and forearms). In some cases, myoclonic twitches were not visually noticeable and were detected only by palpation. In 45.5% of patients, active myoclonic “nodding” was observed - patients made a sharp nodding movement, often combined with trunk propulsion. With MP in the legs, patients felt something like “knocks under the knees”; at the same time, patients squatted slightly or suddenly fell on their knees or buttocks (myoclonic-astatic attacks). Consciousness during myoclonic-astatic attacks remained intact (in the absence of absence seizures) and the patients immediately rose after a fall. The frequency of MP and myoclonic-astatic seizures during MAE was extremely high and, in most cases, amounted to 10-30 attacks per day. A clear dependence of MP on the sleep-wake rhythm was revealed, with an increase in the frequency of attacks in the morning after patients awakened. SHGs were observed in all cases. In most cases (63.6%) GSP was the first manifestation of the disease. Characterized by generalized tonic-clonic and clonic-tonic-clonic paroxysms lasting from 30 seconds. up to 2 min. A feature of GSP was the presence of paroxysms with alternating hemiconvulsions: hemiconvulsive attacks that change side from one attack to another. In 36.4% of patients, a high frequency of attacks was observed (from once a day to once a week) and in 63.6% - once a month or less. Just as with MP, an increase in GSP was observed during the period of awakening of patients.
Absence seizures were observed in 72.7% of patients. The disease debuted with absence seizures in 18.2% of patients. Short simple absences predominated, as well as absences with a myoclonic component (myoclonus of the eyelids, cheeks). The frequency of absence seizures was high (20-30 days), reaching a maximum in the morning. Partial seizures were observed in 27.3% of patients. Short simple partial motor seizures were noted, the frequency of which did not exceed 1 time per month. There was a predominance of partial seizures at night.
In a number of patients, factors were identified that significantly provoked the occurrence or increase in frequency of attacks. The main provoking factors were: sleep deprivation (45.5% of patients), sudden forced awakening (27.3%). These factors caused an increase in GSP, myoclonic and myoclonic-astatic seizures, but non-absences and partial paroxysms.
Neurological examination revealed abnormalities in the majority of patients (81.8%). The following were noted: pyramidal hemisyndrome (anisoreflexia, anisotonia in combination with the appearance of pathological reflexes, but in the absence of movement disorders) - 45.5%, coordination disorders (instability in the Romberg position, mild intention tremor and missedness on one side) - 27.3%. More than half of the patients (54.5%) had a decrease in intelligence combined with hyperactive behavior.
During the study, in no case were normal EEG results noted. Characteristic was a slowdown in the main activity of the background recording (54.5%), the appearance of generalized epileptic activity (63.6%) and regional changes (81.8%). Generalized epileptic activity was represented by peak-wave (45.5%) and polypeak-wave complexes (18.2%) with a frequency of 3 Hz. Pronounced amplitude asymmetry of the complexes, while maintaining their bilaterally synchronous nature, was observed in 36.4% of patients. In the majority of patients (81.8%), regional changes in bioelectrical activity were observed. Slowing in the theta rhythm was observed (54.5%), more often in the central parietal leads, as well as regional peak-wave activity in the temporal leads (27.3%). Carrying out a hyperventilation test and rhythmic photostimulation did not significantly change the EEG pattern.
CT and MRI studies revealed changes in 45.5% of patients. The following were observed: small arachnoid cysts on the convexital surface of the brain (18.2%), subatrophy of the cerebral cortex (9.1%), moderate ventriculomegaly (9.1%). In one patient, MRI revealed an anomaly of brain development with hypoplasia of the left hemisphere and asymmetric ventriculomegaly.
The following drugs have been tested in the treatment of MAE: barbiturates, carbamazepines, succinamides, valproate and lamotrigine. With the use of barbiturates, a significant reduction in the frequency of epileptic seizures was achieved in 37.3% of patients, but in all cases there was a sharp increase in hyperactivity in children. In no case did the administration of carbamazepines have a significant effect on the frequency of attacks. In one patient with absence seizures, the use of carbamazepine resulted in a significant increase in seizures. With monotherapy with valproates (Depakine, Konvulex, apilepsin), a decrease in attacks was observed in 45.5% of cases. The most optimistic results were obtained when using valproic acid derivatives in combination with other drugs. When using valproates (900 - 2000 mg daily) with lamotrigine (25 - 50 mg daily), it was possible to achieve complete therapeutic remission in 30.0% of patients and significant improvement in 50% (of those taking valproate). The combination of valproates with succinamides or benzodiazepines caused a marked reduction in the frequency of attacks in the majority of patients (60.0%), but in no case was remission achieved. Thus, in the general group of patients with MAE, complete remission was achieved in 27.3% of cases, a significant reduction in attacks - 63.6%, and no effect - 9.1% (including in patients with brain malformations). Follow-up was followed for 1-3 years.
Discussion
MAE is a rare form of epilepsy and its frequency, according to Doose (Rojer), is 2% among all forms of epilepsy in children. According to the results of our own studies, the frequency of MAE is 5.6% among generalized forms of epilepsy. The taxonomic position of MAE among other epileptic syndromes is not completely clear. The following features are similar to idiopathic forms of epilepsy: the predominantly generalized nature of seizures (myoclonic, myoclonic-astatic and absence seizures), the detection of generalized peak and polypeak wave activity of 3 Hz on the EEG, the positive therapeutic effect of valproic acid drugs. A number of features make MAE similar to symptomatic forms of epilepsy: the possibility of partial seizures, the high frequency of detection of focal neurological symptoms and intellectual impairment, and relative therapeutic resistance. Thus, this syndrome is, as it were, transitional from idiopathic generalized forms of epilepsy (for example, absence forms of epilepsy) to symptomatic forms (Lennox-Gastaut syndrome).
A number of authors question the nosological independence of MAE (Willye). According to Aicardi, MAE is a special (so-called “myoclonic”) variant of LGS. At the same time, accurate diagnosis of these two syndromes is fundamentally important in relation to treatment tactics, course and prognosis of the disease. We present differential diagnostic criteria for MAE with LGS. The cardinal difference between the clinical manifestations of these two syndromes is the presence of myoclonic-astatic seizures in MAE and atonic-astatic seizures in LGS. This difference is especially noticeable during video monitoring of seizures. As a rule, myoclonic-astatic seizures begin with short myoclonic jerks of the lower extremities, after which the patient falls to his knees or buttocks. With atonic-astatic attacks, there is a sudden decrease in muscle tone and a vertical fall in patients with severe trauma. The possibility of absence seizures in both forms of epilepsy was indicated by a number of authors (Guerrini). The different nature of absence seizures is emphasized: typical (frequency of peak-wave complexes 3 or more Hz) with MAE (Beck-Mannagetta) and atypical (complexes acute - slow wave of low frequency - less than 2.5 Hz) absences - with LSH (Zifkin). notes in LSH the absence of a direct connection between the occurrence of epileptiform EEG patterns and the appearance of clinical paroxysms. At the same time, in most cases with MAE there is a clear relationship between the appearance of peak- and polypeak-wave complexes and the occurrence of absence seizures and myoclonic twitches. In addition, typical for LSH is a pronounced slowdown in the main activity of the background EEG recording and the appearance of acute-slow wave complexes, both generalized and multifocal. These changes are not typical for MAE. One of the important diagnostic signs for LGS is the occurrence before the onset of attacks or immediately after their onset of a rapid decline in intelligence.
Thus, the study showed the clinical and electroneurophysiological uniqueness of MAE, which indicates, in our opinion, the nosological independence of this syndrome. According to the results obtained, the criteria for the diagnosis of MAE are:
onset of seizures in preschool age (maximum 2-3 years),
myoclonic and myoclonic-astatic seizures as the main type of paroxysms, a high frequency of the presence of GSP, from which the disease often debuts, the possibility of partial seizures, a high frequency of detection of neurological disorders and decreased intelligence, relative resistance to monotherapy with basic anticonvulsants.
The presented diagnostic criteria will allow us to distinguish MAE from other myoclonic forms of epilepsy in children and prescribe adequate anticonvulsant therapy. The optimal therapeutic regimen is a combination of valproate with lamictal or succinamide or benzodiazepines.
Bibliographic index
Zifkin BG - “The Lennox - Gastaut syndrome.” - In: Comprehensive epileptology, - eds. M.Dam & L.Gram, - 1990, - pp.123-131. New York, Raven Press.
Aicardi J. - “Epilepsy in children.” - 1994. New York, Raven Press.
G.Beck-Mannagetta, C.Dehe-Steffens, B.Schmits, D.Jans.- “Genetic aspects of epilepsies of early childhood with mioclonic - astatic seizures.”- In: Epileptic seizures and syndromes,- eds. P.Wolf, - 1994, -pp. 165-168. London, John Libbey.
R.Guerrini, Ch.Drave, G.Gobbi et al. - “Idiopathic generalized epilepsies with myoclonus in infancy and childhood.” - In: Idiopathic generalized epilepsies, - eds. A. Malafosse, P. Genton, E. Hirsch et al., - 1994, - pp. 267-280. London, John Libbey.
Wyllie E. - “The treatment of epilepsy.” - 1993, - 1238 P. Philadelphia. Lea & Febiger.
J. Roger et al. - “Epileptic syndromes in infancy, childhood, adolescence.” - Paris - 1992-p.316-327 (classification).
H.Jackson.- “On a case of fits resembling those artificially produced in guinea - pigs.”- In: Selected writings of John Hughlings Jackson.- 1931.- V.1.- p. 362. London, Hodder & Stought
Clinical examination of the patient in order to make a diagnosis.
This examination is quite simple. It requires a careful history and repeated video-EEG recordings to demonstrate the presence of MP with generalized SV discharges, spontaneous or those promoted by drowsiness, noise, contact, or IRS. Sleep recordings can show weak activation of discharges and focal disturbances. Neuroimaging is useful to confirm the absence of brain damage, but is not necessary in the presence of typical symptoms. A neuropsychiatric examination is more useful for checking psychomotor development and tracking the course of the disease.
Treatment
VPA in monotherapy is the drug of choice, its use should be started as early as possible. It is preferable to use a solution rather than a syrup, as it is better tolerated by the child. It is necessary to strictly monitor the concentration of the drug in plasma.
A daily dose of 30 mg/kg 3 times a day is usually sufficient, but a higher dose may be required in some patients. VPA is also effective for possible febrile seizures. If VPA is ineffective, it can be used in combination with a benzoadiazepine (CLB or NTZ) or ETS and reconsider the diagnosis. Treatment should be continued for 3-4 years after the onset of the disease if it is well tolerated. In cases of purely reflex symptoms, drug therapy may not be used. If it has already started, it can be stopped abruptly, but if there is no hypersensitivity. If PEP occurs in a teenager, short-term treatment can be given at that age. Conservative treatment should be combined with psychological assistance to the family.
Treatment of juvenile myoclonic epilepsy
Regular events
Not only pharmacotherapy for epilepsy is important, but also the patient’s compliance with certain life standards to avoid provoking seizures. As with other types of epilepsy, in JME seizures can be caused by a violation of the regime, mental and physical overload, stress, lack of sleep, and drinking alcohol-containing drinks. Therefore, the patient should avoid such provoking factors. A calm, simple and leisurely lifestyle, being in nature, away from the bustle of the city, has a positive effect on the course of the disease. Due to this, some families where a child is diagnosed with JME move and live in rural areas.
Pharmacotherapy
Drug therapy for JME is carried out with valproate. Monotherapy with these drugs turned out to be effective against all types of seizures accompanying the JME clinic - myoclonic, tonic-clonic and absence seizures. If monotherapy is insufficient, combination treatment is possible. Relief of resistant absence seizures is achieved by combining valproate with ethosuximide, and resistant clonic-tonic seizures by combining valproate with primidone or phenobarbital.
Clonazepam is effective for controlling myoclonic paroxysms, but its effect does not extend to tonic-clonic generalized seizures. At the same time, complete relief of myoclonic attacks deprives the patient of the opportunity to know in advance about the approaching tonic-clonic attack by the myoclonic manifestations that appear before it. Therefore, the prescription of clonazepam is justified only for persistent myoclonic paroxysms and should be combined with the drug valproic acid.
The results of treatment of JME with new generation antiepileptic drugs (levetiracetam, lamotrigine, topiramate) are still being tested in clinical settings. Levetiracetam is highly promising.