Юношеская миоклоническая эпилепсия — это форма генерализованной эпилепсии, основу клинической картины которой составляют миоклонические приступы — асинхронные мышечные сокращения, кратковременно возникающие в симметричных участках тела, преимущественно в руках и плечевом поясе. Наряду с миоклоническими эпизодами в клинике могут наблюдаться абсансы и клонико-тонические генерализованные эпиприступы. Юношеская миоклоническая эпилепсия диагностируется на основании клиники заболевания и результатов электроэнцефалографии, при исключении органической церебральной патологии по данным неврологического осмотра и МРТ. Лечение проводится преимущественно препаратами вальпроевой кислоты. Как правило, необходимо пожизненное наблюдение эпилептолога.
История заболевания и номенклатура
Синдром доброкачественной миоклонической эпилепсии у детей (СДМЭД) не имел четкого определения до первого описания его у 7 детей младшего возраста в 1981 г. (Драве и Биор, 1981). В этой статье СДМЭД определялся как случай миоклонических припадков (МП) без припадков другого типа, кроме простых фебрильных припадков (ФП), в первые три года жизни у здоровых детей. Эти МП легко поддавались лечению и становились достаточно редкими в течение дальнейших лет периода детского возраста. Психомоторное развитие оставалось нормальным, каких-либо тяжелых психологических последствий не наблюдалось. С тех пор в литературе было описано много других случаев. СДМЭД включен в Международную классификацию 1989 г. среди генерализованных идиопатических эпилепсий (Комиссия, 1989). Некоторые авторы описали у больных рефлекторный МП, вызываемый шумом или разговором, и предложили различать две отдельные нозологические формы, причем 2-я была названа рефлекторной миоклонической эпилепсией у детей (Риччи и др., 1985). Мы думаем, что такое деление нецелесообразно и будем описывать все случаи как СДМЭД.
Насколько нам известно, в литературе на сегодняшний день описано 98 случаев, из которых 88 соответствуют классическому описанию СДМЭД, а 10 определялись как «рефлекторная СДМЭД».
Сейчас под нашим наблюдением находятся 5 новых больных, четверо из которых со спонтанными и рефлекторными припадками, что доводит общее число больных до 103.
В первом описании СДМЭД указывалось, что болезнь начиналась до 3-летнего возраста, тогда как в последующих сообщениях говорилось о более позднем начале у некоторых больных — до 4 лет 8 месяцев (Джиованарди-Росси и др., 1997). Это означает, что эпилепсия одного типа может появиться в разном возрасте (Гуэрини и др., 1997).
Общие положения
Эпидемиология
Согласно немногочисленным имеющимся в наличии эпидемиологическим данным, СДМЭД составляет менее 1% всех эпилепсий (Центр Св. Павла, 1997), 1,3% и 1,72% эпилепсий, которые начинаются в первый год жизни (Далла Бернардина и др., 1983), и 0,39% эпилепсий, начинающихся в первые 6 лет жизни (Охтсука и др., 1993).
Пол
Распределение по полу: 52 мальчика и 27 девочек.
Генетика
Генетика СДМЭД неизвестна. Больных мало, а случаи семейного СДМЭД описаны не были. В семейном анамнезе эпилепсия или фебрильные припадки (ФП) присутствуют у 39% из 80 больных. У 70 больных заболеваемость ФП в семье составляла 17%, а заболеваемость эпилепсией — 24%. Трудно оценить тип эпилепсии, обнаруживаемый у родственников. В 10 случаях это, вероятно, была идиопатическая эпилепсия. В случае, описанном Арзиманоглоу и др. (1996), пробандом был 2-й из 2 братьев, а старший страдал типичной эпилепсией с миоклонически-астатическими припадками (ЭМАП, синдром Дуза).
Анамнез жизни
В анамнезе большинства больных патологии до начала МП не было. Только у двух (1,9%) были сопутствующие заболевания: синдром Дуза (Драве и др., 1992) и гиперинсулиновый диабет (Коламария и др., 1987).
Однако случаи ФП не являются редкими: 19 больных из 64 (30%). ФП были всегда простыми, однако редкими (1-2) и наблюдались до наступления миоклоний и до начала лечения (15 случаев).
Клинические и ЭЭГ проявления
Возраст, в котором начинается заболевание, — обычно от 4 месяцев до 3 лет. Редко начало более раннее. О более позднем начале сообщили Джиованарди-Росси и др. (1997) — 4 года 9 месяцев — и Лин и др. (1998) — 3 года 9 месяцев.
Вначале МП бывают короткими, чаще всего редкими, с вовлечением верхних конечностей и головы, редко — нижних конечностей. У младенцев они едва заметны, и родителям иногда бывает трудно определить их начало и частоту. Они часто говорят о «спазмах» или «кивании головой». Позже частота приступов возрастает.
Записи видео-ЭЭГ позволили нам провести точный анализ этих приступов. Это более или менее массивные миоклонические подергивания, поражающие туловище и конечности, провоцирующие наклон головы и движение верхних конечностей вверх и кнаружи со сгибанием нижних конечностей, а иногда — вращением глазных яблок. Их интенсивность у каждого ребенка своя, у одного и того же пациента она может меняться при каждом приступе. При тяжелых формах отмечается резкое выпадение предметов из рук, иногда больной падает. При формах средней тяжести отмечается только короткое движение головы вперед или даже простое закрытие глаз, как правило, припадки очень короткие (1-3 сек.), хотя они могут длиться и дольше (у детей старшего возраста) и состоять из псевдоритмичных подергиваний, длящихся не более 5-10 сек. Они развиваются несколько раз в день через разные промежутки времени. В отличие от детских спазмов, они не протекают длинными сериями и развиваются не в момент пробуждения, а, скорее, при дремоте. У некоторых больных вызвать припадки может резкий шум, внезапный контакт или повторная фотостимуляция (ИФС). При единичных припадках оценить состояние сознания трудно. Только при повторных припадках наблюдается легкое нарушение сознания без прерывания деятельности. Мы не наблюдали внезапных коротких вокализмов, о которых сообщает Лин и др. (1998). Авторы указывают на вовлечение в патологический процесс диафрагмы и / или абдоминальных мышц, вызывающих экспираторный шум. При рефлекторном МП миоклонус может быть вызван как в состоянии бодрствования, так и сна, порог возбудимости низкий на стадии 1, но он постепенно повышается во время медленных стадий (Риччи и др., 1995). У больных с рефлекторным МП фаза быстрого сна не записывалась и не анализировалась. Т.к. развитие ребенка идет нормально, родители и педиатры принимают эти движения за патологию.
Картина ЭЭГ вне приступа в пределах нормы.
Миоклонии всегда связаны с ЭЭГ разрядом. Записи показывают, что миоклонии сопровождаются разрядом быстрых генерализованных спайк-волн (СВ) или полиспайк-волн (ПСВ) частотой более чем 3 Гц, с той же продолжительностью, что и миоклонии. Это более или менее регулярный разряд, и он может начинаться в лобных областях и в темени. Миоклонии короткие (1-3 сек.) и обычно изолированные. За миоклоническим подергиванием может последовать короткая атония. Иногда после приступа появляются произвольные движения, расцениваемые как нормальное сокращение мышц. И только у одного больного мы отметили сочетание миоклоний в дельтовидной мышце с чистой атонией шейных мышц. Во время сонливости миоклонии усиливаются, но не всегда. Они исчезают во время медленного сна. МП, вызванные тактильными и слуховыми раздражителями, имеют одни и те же характеристики. Риччи и др. (1995) указывают на то, что первичные проявления обычно, но не всегда, проявляются только симптомом моргания, за которым через 40-80 мсек. следует миоклоническое подергивание руки. После миоклонического приступа наступает рефрактерный период, длящийся от 20-30 сек. до 1-2 мин, во время которого внешние стимулы, даже испуг, не провоцируют приступы. Повторная фотостимуляция (ПФ) тоже может спровоцировать МП (Драве и др., 1992; Тодт и Мюллер, 1992; Джиованарджи-Росси и др., 1997; Лин и др., 1998).
Данные интериктальной ЭЭГ без патологии. Спонтанные разряды СВ редки; в центральных областях мозга можно обнаружить медленные волны. ПФ вызывает СВ без имеющейся сопутствующей миоклонии. Записи короткого дневного сна показали нормальную организацию сна, генерализованные разряды СВ могут появляться во время быстрого сна.
Юношеская миоклоническая эпилепсия — симптомы и лечение
Основной диагностический критерий заболевания — это наличие миоклонических приступов.
Сбор анамнеза
На приёме врач спрашивает о необычных внезапных состояниях:
- вздрагиваниях в теле;
- дежавю — состоянии, при котором человек ощущает, что когда-то уже был в подобной ситуации или месте;
- потере сознания и т. д.
Пациенты могут не обращать внимания на такие симптомы и считать их своей особенностью. Абсансы и генерализованные тонико-клонические приступы с потерей сознания, особенно во сне, они могут и вовсе забывать. Поэтому при сборе анамнеза важно выяснить обстоятельства приступа не только у самих пациентов, но и у родственников и очевидцев.
Электроэнцефалограмма (ЭЭГ)
Основным способом диагностики эпилепсии является электроэнцефалограмма — метод исследования, при котором регистрируется суммарная электрическая активность клеток коры головного мозга.
Сейчас диагноз «эпилепсия» устанавливают с помощью длительного видео-ЭЭГ мониторинга — электроэнцефалограмма записывается параллельно с одной или несколькими видеокамерами, датчиком ЭКГ и при необходимости дополнительным контролем мышечной активности, частоты и глубины дыхания.
Основной фон биоэлектрической активности при юношеской миоклонической эпилепсии, как правило, соответствует возрастной норме. Патологическая активность проявляется короткими и генерализованными разрядами полиспайков (островолновых комплексов), которые регистрируются при миоклонических вздрагиваниях и полипик-волновыми комплексами между приступами.
При заболевании часто встречается феномен фотосенситивности. Для её выявления во время ЭЭГ пациента просят закрыть глаза и проводят ритмичную фотостимуляцию с частотой около 15 Гц [16].
Эпилептическая фотосенситивность — это предрасположенность к приступам под влиянием света. Может протекать бессимптомно или проявляться эпилептическими приступами под воздействием провоцирующих факторов: видеоигр, работы за компьютером, просмотра телевизора, мигающего освещения в ночных клубах и света природного происхождения.
На МРТ патологические изменения в головном мозге при юношеской миоклонической эпилепсии не выявляются [17].
Интеллект и неврологический статус при заболевании находятся в норме. Выражена эмоциональная неустойчивость и признаки невротического развития личности: резкая смена настроения, вспыльчивость и повышенная тревожность
Течение и лечение
У детей с СДМЭД не наблюдаются припадки какого-либо другого типа, даже если они остаются без лечения (до 8,5 лет у одного нашего больного), особенно это касается малых эпилептических или тонических припадков. Результаты клинического обследования нормальные. Интериктальный миоклонус был описан только Джиованарди-Росси и др. (1997) у 6 больных. Анализируя состояние наших больных, мы обнаружили легкий интериктальный миоклонус у 2 по записям ЭЭГ. Многие больные не были обследованы, но когда КТ и МРТ были выполнены, результаты оказались нормальными (33 больных).
Исход, вероятно, зависит от раннего диагноза и лечения. Миоклонии легко поддаются монотерапии вальпроатом, развитие ребенка при этом соответствует возрасту. При отсутствии лечения у больного продолжаются миоклонические припадки, что может привести к нарушению психомоторного развития и поведенческим отклонениям.
Более или менее досконально способы лечения апробированы на 74 больных. У 65 больных проводилась монотерапия, у 6 — политерапия, у 3 лечение не проводилось. Монотерапия включала вальпроат (ВПА), фенобарбитал (ФБ), нитразепам (НТЗ). 6 больных получали терапию примидоном (ПРМ) и этосуксимидом (ЭСМ). В результате лечения припадки исчезли у 69 больных (93%).
Эти данные подтверждают, что при СДМЭД ВПА является препаратом первого выбора. Однако лечение должно проходить под контролем концентрации препарата в плазме, т.к. неправильный прием может привести к рецидиву или вызвать резистентную к препаратам эпилепсию.
Отдаленные результаты и прогноз
Длительность наблюдения за 63 больными составила от 9 мес. до 27 лет, из них за 45 пациентами наблюдали около 5 лет. Возраст больных в период наблюдения — от младше 5 до старше 15 лет.
Во всех случаях МП купировались. Длительность заболевания известна у 52 больных: у большинства из них МП длились менее одного года, у 7 — от 1 до 2 лет, только у 5 — более двух лет.
О возникновении генерализованных эпилептических припадков (ГЭП) после прекращения МП сообщалось в случае 10 больных без сопутствующих МП.
Было доложено о результатах наблюдения 74 больных.
Приступы прекратились у 28 больных в возрасте старше 6 лет. У 6 детей этого же возраста приступы сохраняются: при фотостимуляции — у 2 (Драве и др., 1992), по неизвестной причине — у 3 (Джиованарди-Росси и др., 1997). Также приступы сохраняются у 13 больных младше 6 лет. 3 больных младше 6 лет остались без лечения (Риччи и др., 1995). О 24 больных информации нет.
Картина ЭЭГ известна для 55 больных. Она быстро нормализовалась у 23. Редкие спонтанные генерализованные СВ сохранялись у 13. Фотосенситивные изменения наблюдались у 6. Интересно, что фотосенситивность может появиться после исчезновения МП и персистировать долгие годы после прекращения МП вплоть до взрослого возраста. Были также представлены фокальные нарушения. Они были записаны у 5 больных во время пробуждения как СВ в двух лобно-центральных и теменных областях, иногда в лобно-теменной и лобно-височной. Они исчезали с течением времени. Напротив, у других больных они появлялись только во время сна. Они еще сохранялись в конце периода наблюдения у 5 наших больных, и только во время сна.
В целом психологический исход благоприятный, и большинство больных выздоровели. Это точно известно в 69 случаях. 57 (83%) были здоровыми, из них 38 в возрасте 5 лет и старше. У 10 (14%) была легкая заторможенность, они посещали специализированную школу, но никто из них не был помещен в больницу. У 2 больных (3%) была нарушена познавательная способность и выявлены отклонения в формировании личности: у одного пациента был синдром Дауна и сильная чувствительность, у другого до 5 лет были МП с чувствительностью к ИРС и к закрытию глаза. Это психотическое нарушение появилось у него в возрасте 10 лет и прогрессировало.
Этот психологический исход частично зависит от ранней диагностики, позволяющей провести соответствующее лечение и убедить семью в хорошем будущем.
Эти данные подтверждают обычно хороший прогноз СДМЭД. Приступы, вызванные шумом или разговором, было легче контролировать, чем спонтанные. Фотосенситивность же было труднее контролировать, она сохранялась на протяжении нескольких лет после прекращения припадков.
Эпилепсия была известна еще в древнем Вавилоне (т.н. «падучая» болезнь), а Гиппократ описывал ее как болезнь мозга. Небезызвестный Джон Хьюлингс Джексон более 100 лет тому назад описывал эпилепсию, как «периодически возникающие излишние и беспорядочные разряды нервной ткани». В наше время эпилепсией называют состояния, характеризующиеся повторными относительно стереотипными припадками [1].
Эпилепсия (а вернее — эпилепсии) представляет собой группу болезней, при которых основными проявлениями являются повторные пароксизмальные приступы. Распространенность (встречаемость) эпилепсии среди детей несомненно высока, но точная статистика зависит от того, включают ли в число установленных диагнозов случаи, т.н. единичных (изолированных) судорог или бессудорожных приступов, а также фебрильные судороги. Например, в соответствии с данными Millichap J.G. (1968), на долю фебрильных судорог приходится до 2% от всех заболеваний в детском возрасте [2]. В любом случае, эпилепсия является самым частым из серьезных пароксизмальных расстройств функций мозга с распространенностью в общей популяции от 0,3% до 2%, а у детей около 1% [3,4,5].
Этиология и патогенез
В возникновении эпилептических пароксизмов у детей, как и у взрослых, основное значение придается наследственному или приобретенному предрасположению, кроме того, определенную роль играют всевозможные экзогенные воздействия (черепно-мозговые травмы, вирусные и бактериальные инфекции и т.д.).
Собственно эпилептическая активность обусловлена нарушениями метаболизма в синапсах и митохондриях, приводящими к т.н. «эпилептизации» нейронов в различных (наиболее к этому предрасположенных) структурах головного мозга посредством как синоптической, так и несинаптической активации с вовлечением в патологический процесс новых образований центральной нервной системы [3,4].
Классификация эпилепсии
В настоящее время эпилепсии у детей подразделяются в соответствии с классификацией, принятой в 1981 году в г. Киото (Япония). Среди них выделяют: I. Парциальные (фокальные, локальные) припадки: А) простые парциальные; В) сложные парциальные; С) Парциальные с вторичной генерализацией; II. Генерализованные припадки: А) абсансы (типичные и атипичные); В) миоклонические припадки; С) клонические припадки; D) тонические припадки; E) тонико-клонические припадки; F) атонические припадки; III. Неклассифицируемые эпилептические припадки. Menkes J.H. и Sankar R. предлагают различать первичную (идиопатическую), вторичную (симптоматическую) и реактивную эпилепсии. Для всех перечисленных эпилепсий могут быть характерны генерализованные или парциальные (фокальные) приступы [1].
Классификация эпилепсий, эпилептических синдромов и схожих заболеваний, принятая Международной лигой по борьбе с эпилепсией (1989, г. Нью-Дели, Индия) предусматривает следующие рубрики: 1) локализационно-обусловленные формы (очаговые, фокальные, локальтные, парциальные): 1.1. идиопатические (с возрастзависимым началом) — доброкачественная эпилепсия детского возраста с центрально-височными пиками (Роландическая), эпилепсия детского возраста с затылочными пароксизмами, первичная эпилепсия чтения; 1.2. симптоматические — хроническая прогрессирующая парциальная эпилепсия Кожевникова, приступы, характеризующиеся специфическими способами провокации, другие формы эпилепсии с известной этиологией или органическими изменения в мозге (лобная, височная, теменная, затылочная); 1.3. криптогенные; 2) генерализованные формы эпилепсии: 2.1. идиопатические (с возрастзависимым началом) — доброкачественные семейные судороги новорожденных, доброкачественные судороги новорожденных, доброкачественная миоклоническая эпилепсия младенческого возраста, абсансная эпилепсия детская (пикнолепсия), абсансная эпилепсия юношеская, юношеская миоклоническая эпилепсия, эпилепсия с генерализованными судорожными приступами пробуждения, другие идиопатические генерализованные формы эпилепсии, не названные выше, формы, характеризующиеся специфическими способами провокации (чаще — фотосенситивная эпилепсия); 2.2 криптогенные и/или симптоматические — синдром Уэста (инфантильные спазмы), синдром Леннокса-Гасто, эпилепсия с миоклонически-астатическими приступами, эпилепсия с миоклоническими абсансами; 2.3. симптоматические: 2.3.1. неспецифической этиологии — ранняя миоклоническая энцефалопатия, ранняя младенческая эпилептическая энцефалопатия с паттерном «вспышка-угнетение» на ЭЭГ (синдром Отахара), другие симптоматические генерализованные формы эпилепсии, не названные выше; 2.3.2. специфические синдромы; 3) эпилепсии, не имеющие четкой классификации как парциальные или генерализованные: 3.1. имеющие как генерализованные, так и парциальные проявления — судороги новорожденных, тяжелая миоклоническая эпилепсия раннего детского возраста, эпилепсия с непрерывными пик-волнами во время медленного сна, приобретенная эпилептическая афазия (синдром Ландау-Клеффнера), другие неклассифицируемые формы эпилепсии, не определенные выше; 3.2. приступы, не имеющие четких генерализованных или парциальных признаков; 4) специфические синдромы: 4.1. ситуационно обусловленные приступы — фебрильные судороги, приступы, возникающие только в результате острых метаболических или токсических нарушений; 4.2. изолированные приступы или изолированный эпилептический статус.
Клиническая картина
В связи многочисленностью и многообразием известных на сегодняшний день форм эпилепсии, остановимся лишь на некоторых особенностях эпилептических припадков у детей. Отметим при этом, что у детей первого года жизни в силу преимущественного функционирования стволовых структур мозга обычно отмечаются тонические припадки, а клонический компонент приступа формируется в более старшем возрасте.
При генерализованных припадках ребенок, обычно, внезапно теряет сознание и падает, издает стон или крик. Наступает тоническая фаза припадка, когда отмечается резкое напряжение всей мускулатуры: запрокидывание головы назад, цианоз лица с гримасой страха, расширение зрачков с отсутствием реакции на свет, сжимание челюстей, сгибание рук в локтевых суставах, сжимание кулаков, вытягивание нижних конечностей. Далее следует клоническая фаза: судороги верхних и нижних конечностей, головы, иногда происходит непроизвольное мочеиспускание и дефекация. Постепенно судороги урезаются с уменьшением выраженности (мускулатура расслабляется, снижаются сухожильные рефлексы). Ребенок перестает реагировать на окружающую обстановку, засыпает (или приходит в сознание в состоянии полной амнезии).
Абсансы (малые припадки) проявляются кратковременной (несколько секунд) потерей сознания с последующей амнезией. При этом судороги и другие двигательные нарушения могут отсутствовать, хотя нередко наблюдаются моторные феномены различной продолжительности, отдельные миоклонические судороги, элементарные автоматизмы, вегето-висцеральные или вазомоторные нарушения.
Миоклонические судороги (инфантильные спазмы) характеризуются наличием своеобразных спазмов (преимущественно сгибательного типа), прогрессирующим течением, а также специфическими изменениями на ЭЭГ (гипсаритмия).
Проявления парциальных приступов у детей зависят от сохранности или потери сознания во время припадка (простые парциальные без нарушения сознания или сложные парциальные в виде джексоновских, адверсивных и т.д. при нарушениях сознания).
Диагностика
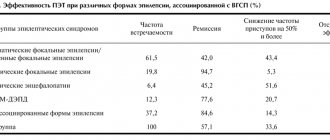
Современные методы диагностики эпилепсии у детей и подростков предполагают широкое использование таких методов, как электроэнцефалография (ЭЭГ), компьютерная томография (КТ), магнитно-резонансная томография (МРТ). При необходимости применяют ЭЭГ-видеомониторинг, позитронно-эмиссионную томографию (ПЭТ), а также целый ряд биохимических иммунологических и прочих специальных методов исследования.
Лечение эпилепсии у детей
Общие принципы лечения эпилепсии включают: 1) соблюдение общего и пищевого режима, 2) терапию антиконвульсантами и дополнительное индивидуальное медикаментозное лечение, 3) психотерапию.
Принципы назначения антиконвульсантов детям с эпилепсией состоят в следующем [1]:
- Выбор препарата определяется типом приступов и потенциальной токсичностью препарата.
- Лечение следует начинать с одного препарата, увеличивая его дозировку до тех пор, пока приступы не прекратятся (или не проявится токсическое действие препарата). Если назначенное средство не приводит к контролю над приступами, его постепенно отменяют (по мере назначения другого препарата и увеличения его дозы).
Концепция монотерапии эпилепсии (т.е. преимущественное использование одного антиконвульсанта для лечения приступов) получила во всем мире широкую поддержку. Она подкреплена результатами многочисленных клинических исследований, в которых задокументированы различные «погрешности» политерапии. Во-первых, хроническая лекарственная токсичность напрямую связана с числом препаратов, потребляемых ребенком. Даже если ни один из этих препаратов не назначается в дозировке, могущей быть токсичным, их влияние на сенсорные и интеллектуальные функции, оказывается кумулятивным. Во-вторых, политерапия может приводить к аггравации приступов у значительной части пациентов (особенно детского возраста). В-третьих, при политерапии бывает сложно (или невозможно) идентифицировать причину побочных реакций.Reynolds E.H. и Shovron S.D. (1981) указывают, что переход с политерапии на монотерапию сопровождается лучшим контролем над судорогами и улучшением показателей интеллектуального развития (когнитивных функций) [6]. Hanson R.A. и Menkes J.H. еще 30 лет назад провели сравнительный анализ эффективности моно- и политерапии. Оказалось, что только в четверти случаев одного антиконвульсанта было недостаточно для контроля над судорогами и требовалось назначение второго препарата [7].
- Изменения в дозе препарата (препаратов) следует осуществлять постепенно, обычно не чаще, чем раз в 5-7 дней.
- Маловероятно, что контроль над приступами будет достигнут при использовании менее известного средства, если препарат первого выбора не подошел. В то же время вероятность необычных побочных реакций при назначении нового или редко используемого препарата весьма велика.
- Добившись контроля над приступами, следует придерживаться использования назначенного препарата в течение длительного времени.
- У пациентов, принимающих фенобарбитал, фенитоин, карбамазепин и этосуксимид, чрезвычайно важны уровни антиконвульсантов в крови. Эти концентрации следует часто мониторировать, особенно у пациентов, не отвечающих на проводимую терапию или демонстрирующих признаки лекарственной интоксикации.
- Известно, что на фоне лечения антиконвульсантами нередко развиваются нарушения со стороны крови и функции печени. Им предшествует т.н. асимптоматическая фаза, которую можно выявить лабораторными и инструментальными методами. В этой связи предлагается проведение рутинных исследований крови и оценка состояния печени. В то же время некоторые американские исследователи указывают, что если каждого пациента, получающего антиконвульсанты, обследовать в полном объеме, то стоимость терапии эпилепсии станет поистине астрономической [8].
- Антиконвульсанты следует отменять с осторожностью и постепенно. Резкая отмена препарата, особенно, если речь идет о барбитуратах, является наиболее частой причиной развития эпилептического статуса [1].
Ниже в алфавитном порядке приведены сведения об основных препаратах, используемых при лечении эпилептических синдромов у детей.
Адренокортикотропный гормон (АКТГ, кортикотропин, адренокортикотропин, АКТГ-гель, синактен-депо)
Показания: препарат 1-го выбора при лечении инфантильных спазмов (синдром Уэста) и других младенческих эпилептических энцефалопатий, препарат 2-го выбора при терапии синдрома Леннокс-Гасто, детской формы кожевниковской эпилепсии, эпилептической афазии (синдром Ландау-Клеффнера), миоклонико-астатической эпилепсии.
Дозировка: в течение 1 недели лечения — 150 Ед/кв.м, 2 — 75 Ед/кв.м, 3-4 — 75 Ед/кв.м, 5-6 — 50 Ед/кв.м, 7-8 — 40 Ед/кв.м, 9-10 — 20 Ед/кв.м, 11-12 — 10 Eд/кв.м поверхности тела через день.
Ацетазоламид
Показания: при больших, малых и парциальных комплексных приступах, при катамниальных припадках в качестве препарата 2 выбора.
Дозировка: 10-15 мг/кг/сутки (в 1-3 приема).
Примечание: принимать ацетазоламид следует с препаратами калия.
Бензобарбитал
Показания: при генерализованных тонико-клонических, миоклонических, простых парциальных и психомоторных припадках.
Дозировка: (3-6 лет) 0,1-0,15 г/сутки, (7-10 лет) 0,3-0,5 г/cутки, (11-14 лет) 0,3-0,4 г/сутки (принимать 2-3 раза в день).
Бромиды/брома соли (натрия бромид, калия бромид)
Показания: как препарат 3 выбора при терапии больших, реже — фокальных припадков.
Дозировка: 75 мг/кг/сутки (в 1-2 приема).
Примечание: бромиды являются препаратами 1 выбора при лечении интермиттирующей порфирии.
Вальпроевая кислота (депакин, депакин-хроно, депакин-энтерик, апилепсин, ацедипрол, конвулекс, конвульсофин, орфирил, энкорат)
Показания: пикнолептические абсансы, миоклонико-астатические, генерализованные фотосенситивные, миоклонические, большие тонико-клонические приступы, инфантильные спазмы, простые и комплексные парциальные припадки. Дозировка: 30-40 мг/кг/сутки Депакин-хроно — 1-2 раза в сутки. Остальные препараты — 2-3 раза в сутки.
Примечание: при необходимости доза может быть увеличена.
Вигабатрин
Показания: резистентные к другим антиконвульсантам парциальные и вторично генерализованные пароксизмы, синдром Уэста, синдром Леннокса-Гасто.
Дозировка: от 40 до 100 мг/кг/сутки (в 1-2 приема).
Примечание: препарат неэффективен при миоклонических приступах и абсансах. Чаще применяется в составе политерапии.
Габапентин
Показания: (для детей старше 12 лет) резистентные (к другим антиконвульсантам) формы парциальных и вторично-генерализованных эпилептических припадков, синдром Леннокса-Гасто (в качестве политерапии).
Дозировка: 10-30 мг/кг/сутки.
Примечание: В РФ препарат не зарегистрирован.
Диазепам (седуксен, реланиум, сибазон, апаурин, валиум, диазепекс, диапам, дикам, калмпоуз)
Показания: при всех формах эпилептического статуса (в/в и per rectum), а также per os кратковременно при лечении фебрильных судорог, учащении припадков (1-2 раза в день).
Дозировка: при лечении эпилептического статуса в/в или ректально — детям 1 года жизни 0,4 мг/кг/сутки, детям старше 1 года — 0,3 мг/кг/сутки. В составе политерапии в возрасте 1-3 года — 2-3 мг/сутки, 3-7 лет — 5-7 мг/сутки, 7 лет и старше — 8-16 мг/сутки.
Карбамазепин
Показания: генерализованные тонико-клонические, простые парциальные, комплексные, вторично генерализованные, психомоторные припадки.
Дозировка: 30 мг/кг/сутки (в 1-3 приема).
Клоназепам
Показания: при всех формах эпилептического статуса (в/в и per rectum), в качестве дополнительного препарата при лечении генерализованной эпилепсии с миоклонико-астатическими припадками, при миоклонических, простых и комплексных парциальных припадках per os (в 1-2 приема).
Дозировка: при лечении эпилептического статуса 0,25-0,5 мг за 30 секунд, при пероральном приеме — 0,15 мг/кг/сутки.
Ламотриджин
Показания: лечение парциальных, комплексных парциальных, вторично-генерализованных и первично генерализованных припадков, атонических приступов, абсансов, резистентных к терапии другими антиконвульсантами, а также синдрома Леннокса-Гасто (дети старше 2 лет).
Дозировка: при терапии ламотриджином и вальпроатом натрия в сочетании с другими противоэпилептическими препаратами или без них: 1-2 недели — 0,15 мг/кг/сутки, 3-4 недели — 0,3 мг/кг/сутки (давать в 1 прием), для достижения терапевтического эффекта дозу увеличивают на 0,3 мг/кг/сутки через каждые 1-2 недели до поддерживающей дозы 1-5 мг/кг/сутки, но не более 200 мг/сутки (в 1-2 приема); при терапии ламотриджином и противоэпилептическими препаратами, инициирующими печеночные ферменты, в сочетании с другими противоэпилептическими препаратами или без них (за исключением вальпроата натрия): 1-2 недели — 0,6 мг/кг/сутки, 3-4 недели — 1,2 мг/кг/сутки (в 2 приема), для достижения терапевтического эффекта дозу увеличивают на 1,2 мг/кг/сутки через каждые 1-2 недели до поддерживающей дозы 5-15 мг/кг/сутки (в 2 приема), но не более 400 мг/сутки.
Примечания: используется при проведении политерапии при неэффективности препаратов первого выбора (детям старше 2 лет).
Мидазолам
Показания: при лечении эпилептического статуса (всех форм), в качестве дополнительного препарата при всех формах приступов (особенно — при миоклонических припадках).
Дозировка: при назначении per os, per rectum — (до 12-летнего возраста) — 7-15 мг/cутки, (после 12-летнего возраста) — 15-45 мг/сутки.
Примечание: при лечении эпилептического статуса у детей — по 0,15-0,3 мг/кг (внутримышечно или ректально).
Нитразепам (радедорм, нитразепам, эуноктин, берлидорм, нитразадон, нитрам, нитросан)
Показания: в качестве дополнительного средства при (политерапии) генерализованной эпилепсии с миоклонико-астатическими приступами, при миоклонических, простых и комплексных парциальных припадках.
Дозировка: 0,4-0,5 мг/кг/сутки (в 1-2 приема).
Примидон (гексамидин)
Показания: при лечении генерализованных тонико-клонических, миоклонических, психомоторных и простых парциальных приступов.
Дозировка: 20 мг/кг/сутки (в 1-2 приема).
Тиагабин (габитрил)
Показания: простые парциальные, комплексные, вторично-генерализованные, психомоторные эпилептические приступы.
Дозировка: 0,5-1,0 мг/кг/сутки.
Примечание: в РФ препарат не зарегистрирован.
Топирамат
Показания: (детям старше 12 лет) при лечении простых и комплексных приступов (с генерализацией и без таковой), больших тонико-клонических припадков, астатических приступов при синдроме Леннокса-Гасто, в качестве дополнительного препарата при терапии резистентных эпилептических синдромов.
Дозировка: 200-400 мг/cутки (в 2 приема).
Фенитоин (дифенин)
Показания: при терапии генерализованных тонико-клонических, простых и комплексных парциальных, а также психомоторных припадков.
Дозировка: 7 мг/кг/сутки (в 1-2 приема).
Примечание: не рекомендуется вводить в одной дозе с другими препаратами (во избежание преципитации).
Фенобарбитал (люминал, смесь Сирейского, глюферал, паглюферал 1, паглюферал 2, паглюферал 3)
Показания: при лечении генерализованных тонико-клонических эпилептических приступов, простых парциальных и психомоторных припадков.
Дозировка: новорожденные и грудные дети — 10-15 мг/кг/сутки, дети старше 1 года — 3-5 мг/кг/сутки (в 1-2 приема).
Примечание: у детей раннего возраста препарат характеризуется выраженной рахитогенной активностью.
Этосуксимид
Показания: при лечении пикнолептических абсансов, миоклонико-астатических и миоклонических припадков.
Дозировка: 30 мг/кг/сутки (в 1-2 приема).
Примечание: начальная доза этосуксимида составляет 10-15 мг/кг/сутки в течение первой недели лечения с последующим повышением дозы до терапевтической [3,9,10].
Немедикаментозное лечение эпилептических синдромов сводится к назначению кетогенной диеты или, в ряде случаев, к проведению нейрохирургических операций.
Несмотря на появление новых, высокоэффективных антиконвульсантов, кетогенная диета, основанная на воспроизведении состояний кетоза и ацидоза при голодании, остается весьма привлекательной альтернативой медикаментозному лечению при лечении синдрома Леннокса-Гасто и других рефрактерных эпилептических синдромов. Механизм контроля за припадками при использовании данного метода диетотерапии остается неясным. Тем не менее, в 90-х гг. прошлого века популярность кетогенных диет в ряде стран значительно возросла [11]. Указанная диета считается особенно эффективной при лечении детей в возрасте 2-5 лет с малыми моторными припадками (у детей более старшего возраста сложно поддерживать состояние кетоза) [1]. К сожалению, в России в детской неврологии кетогенные диеты пока еще не нашли применения.
Не используются широко в нашей стране и нейрохирургические методы лечения эпилепсии. Тем не менее, имеются данные, свидетельствующие об эффективности гемисфероэктомии при синдроме Расмуссена (хронический фокальный энцефалит) [1]. В современной мировой литературе также сообщается об использовании при различных резистентных к лечению эпилептических синдромах следующих видов нейрохирургических операций: передняя темпоральная лобэктомия [12], ограниченная темпоральная резекция [13] и экстратемпоральная неокортикальная резекция [14]. Отдельное место среди хирургических методов лечения занимает стимуляция блуждающего нерва [15].
Дифференциальный диагноз
Если миоклонии начинаются в первый год жизни, их надо дифференцировать с криптогенными детскими судорогами (ДС). ДС клинически отличаются от доброкачественных миоклоний: они более тяжело протекают, включают в себя тонические судороги во всем теле, что никогда не наблюдается при СДМЭД; единичные судороги всегда сочетаются с серийными приступами у данного ребенка; возникновение долгих серийных спазмов происходит после пробуждения. Затем СД на ЭЭГ демонстрируют типичную картину коротких тонических сокращений, которую хорошо описали Руско и Вигевано (1993); редко имеют место длительные миоклонии. Иктальная ЭЭГ переменная: внезапные прерывистые гипсаритмии выравниваются наложенными быстрыми ритмами, большой медленной волной, за которой следует выравнивание или отсутствие видимых изменений. Наступление ДС связано с поведенческими изменениями, затруднением речевого контакта, замедлением психомоторных достижений.
На интериктальной ЭЭГ всегда есть патологические признаки: истинная гипсаритмия, или видоизмененная гипсаритмия, или фокальные нарушения, на них не определяются отдельные или короткие всплески билатеральных синхронных СВ или СДМЭД.
Если психомоторное развитие и ЭЭГ в пределах нормы по результатам нескольких обследований, выполненных во время бодрствования и сна, припадки, похожие на ДС, должны навести на мысль о диагнозе доброкачественного неэпилептического миоклонуса, описанного Ломброзо и Фейерманом (1977). У этих больных даже иктальная ЭЭГ без патологических изменений (Драве и др., 1986; Пашатс и др., 1999).
На первом году жизни может развиваться миоклоническая эпилепсия младенчества, она всегда начинается с длительных и частых фебрильных судорожных припадков, а не с отдельных миоклонических приступов. На втором году психомоторное развитие замедляется.
Если миоклонии начинаются после первого года жизни, можно заподозрить криптогенный синдром Леннокса — Гасто (СЛГ). При СЛГ (Бюмане и Драве, 1992) приступы в основном не миоклонические, а миоклонико-атонические, а чаще тонические, приводящие к внезапным падениям и травмам. Их ЭЭГ картина неоднородна, а в иктальном периоде высокоамплитудная или выравнивается, или определяется высокая медленноволновая активность, за которой следуют низкоамплитудные волны. В самом начале интериктальные ЭЭГ могут быть нормальными, в дальнейшем могут появиться типичные диффузные разряды медленной СВ. Типичные электроклинические признаки во время сна могут быть отложенными по времени. Диагноз основывается на быстрой связи припадков разного типа, таких как атипичные абсансы и аксиальные тонические судороги, на постоянных поведенческих нарушениях поведения и появлении новых навыков и неэффективности антиэпилептических препаратов.
Если миоклонические приступы изолированные или связаны с БЭП, необходимо рассмотреть диагноз миоклонической астазической эпилепсии раннего детского возраста (ЭМАП), хотя при этом синдроме миоклонические астанические припадки редко начинаются до возраста 3 лет (Дуз, 1992). Есть два существенных различия: 1) клинический аспект припадков со ступором, которые не наблюдаются при СДМЭД (Гуеррини и др., 1994); 2) ЭЭГ признаки тоже разные: СВ и ПСВ более многочисленные, сгруппированы в долгие вспышки, связанные типичным тета-ритмом над центро-теменными зонами. Но некоторых больных, вероятно, следовало бы классифицировать как СДМЭД. Подобным образом Дельгадо-Эскуэта и др. (1990) в группу обследованных или больных включили, вероятно, под названием миоклонической эпилепсии детского возраста (МЭРД) случаи как ЭМАП, так и СДМЭД.
Наконец, следует учитывать и другие эпилепсии, начинающиеся в первые три года жизни, при которых миоклонии являются основным типом приступов и которые имеют изменчивый прогноз. Они неоднородны: сочетание приступов других типов, постоянные фокальные нарушения на ЭЭГ, замедленное психомоторное развитие в прошлом, плохая реакция на лечение, неясный прогноз (Драве и др., 1992).
Миоклонически-астатические приступы
Генерализованная эпилепсия с миоклонически-астатическими приступами: диагностика и терапия
К.Ю. Мухин, А.С. Петрухин, Е.А. Рыкова
Миоклонические приступы (МП) являются частым проявлением многих форм эпилепсии детского возраста (Guerrini). В связи с этим, важной задачей является систематизация различных эпилептических синдромов, проявляющихсям иоклоническими приступами. Предпринимались различные подходы к систематизации данных видов приступов. Было предложено выделять симптоматические, криптогенные иидиопатические, а также первично и вторично генерализованные формы миоклонической. В последние годы с внедрением в клиническую практику методов видео-ЭЭГ мониторинга и прижизненной визуализации головного мозга (КТ,МРТ, ПЭТ) появилась возможность более точной дифференцировки различных эпилептических синдромов. Современная классификация эпилепсии(1989) выделяет следующие эпилептические синдромы у детей, проявляющиеся МП:
ранняя миоклоническая энцефалопатия;
доброкачественная и злокачественная (тяжелая) миоклонические эпилепсии младенчества;
эпилепсия с миоклоническими абсансами;
синдром Леннокса-Гасто (СЛГ);
юношеская миоклоническая эпилепсия;
эпилепсия с миоклонически-астатическими приступами (МАЭ).
Первое сообщение о МАЭ было сделано Jackson в 1886году. Автор наблюдал 7 летнего ребенка, «у которого с 2.5 лет наблюдались приступы падений». Лишь в 1964 году Doose представил подробное описаниесиндрома и выделил МАЭ под названием»акинетический миоклонический petit mal». Согласно оригинальному описанию автора, данная форма эпилепсии характеризуется высокой генетической предрасположенностью, дебютом приступов в дошкольном возрасте; проявляется преимущественно миоклоническими и миоклонически-астатическими приступами и имеет неблагоприятный прогноз. Вместе с тем, до настоящего времени дискутируется вопрос о нозологической самостоятельности МАЭ, которая признается не всеми авторами. Отсутствуют четкие критерии диагностики данной формы эпилепсии и, в частности, отличие ее от так называемого»миоклонического варианта» СЛГ. Целью настоящего исследования явилась клиническая систематизация, изучение электронейрофизиологических и нейрорадиологических особенностей МАЭ, а также терапевтической тактики.
Под нашим наблюдением находилось 11 больных МАЭ в возрасте 4-15 лет (средний — 7.4+/-1.5 лет), из них 7мужского и 4 — женского пола. Проводился видеомониторинг приступов, исследование ЭЭГ(Медикор, 18 каналов), ЭЭГ картирование и компьютерная томография головного мозга ( SOMATOM с шагом сканирования 4 и 8мм ). Полученные результаты были обработаны на IBM PC 386 сиспользованием базы данных PARADOX и статистического пакета QUATRO-PRO.
Результаты исследования
Среди обследованных в эпилептическом центре 196 детей с генерализованными формами эпилепсии МАЭ выявлена в 11 случаях, что составляет 5.6%. Согласно данным анамнеза, патология перинатального периода констатировалась у большинства больных(54.5%). Доминировали нарушения в периоде беременности: токсикозы, угроза прерывания беременности ( с приемом гормональных препаратов). В 18.2% случаев отмечались проявления родовой травмы. Семейных случаев эпилепсии выявлено не было. Дебют эпилепсии варьировал от 11 мес. до 5лет, составляя в среднем 2.3+/-1.90 лет. В 81.8% случаев начало заболевания приходилось на возрастной интервал 1-3 года. В подавляющем большинстве случаев (81,8%) МАЭ дебютировала с генерализованных судорожных приступов (ГСП). У одного пациента заболевание начиналось с абсансов и у одного — с тонико-клонических фебрильных пароксизмов. Средний возраст дебюта ГСП составлял 2.4+/-0.38 лет, абсансов — 4.0+/-0.57 и миоклонических приступов -4.0+/-0.40.Клинические проявления МАЭ были полиморфны и включали различные виды приступов: миоклонические, миоклонически-астатические, типичные абсансы, ГСП и парциальные пароксизмы. Ядром» МАЭ являлись миоклонические и миоклонически-астатические приступы. Однако ни в одном случае заболевание не начиналось с данных приступов. МП характеризовались короткими, молниеносными, обычно асинхронными и аритмичными подергиваниями малой амплитуды в ногах и в руках (кисти и предплечья). В некоторых случаях миоклонические подергивания визуально не были заметны и обнаруживались лишь пальпаторно. У 45.5% больных отмечались активные миоклонические «кивки» — пациенты совершали резкое кивательное движение, нередко сочетавшееся с пропульсией туловища. При МП в ногах больные ощущали как бы «удары под колени»; при этом пациенты слегка приседали или внезапно падали на колени или ягодицы (миоклонически-астатические приступы). Сознание при миоклонически-астатических приступах оставалось сохранным (при отсутствии абсансов) и больные мгновенно поднимались после падения. Частота МП и миоклонически-астатических приступов при МАЭ была крайне высока и составляла, в большинстве случаев, 10-30 приступов в сутки. Выявлялась четкая зависимость МП от ритма сон-бодрствование с нарастанием частоты приступов в утренние часы после пробуждения пациентов. ГСП наблюдались во всех случаях. В большинстве случаев (63.6%) ГСП являлись первым проявлением заболевания. Характеризовались генерализованными тонико-клоническими и клонико-тонико-клоническими пароксизмами продолжительностью от 30 сек. до 2 мин. Особенностью ГСП являлось наличие пароксизмов с альтернирующими гемиконвульсиями: гемиконвульсивные приступы, меняющие сторону от одного приступа к другому. У 36.4% больных констатировалась высокая частота приступов (от 1раза в сутки до 1 раза в неделю) и у 63.6% — 1 раз в месяц и реже. Так же, как и при МП, наблюдалось учащение ГСП в период пробуждения пациентов.
Абсансы отмечались у 72.7% больных. Заболевание дебютировало с абсансов у 18.2% пациентов. Преобладали короткие простые абсансы, а также абсансы с миоклоническим компонентом (миоклонии век, щек). Частота абсансов была высокой (20-30 всутки), достигая максимума в утренние часы. Парциальные приступы констатировались у 27.3%пациентов. Отмечались короткие простые парциальные моторные приступы, частота которых не превышала 1 раза в месяц. Наблюдалось преобладание парциальных приступов в ночное время.
У ряда больных были выявлены факторы, в значительной степени провоцирующие возникновение или учащение приступов. Основными провоцирующими факторами были: депривация сна (45.5% пациентов), внезапное насильственное пробуждение (27.3%). Данные факторы вызывали учащение ГСП, миоклонических и миоклонически-астатических приступов, но неабсансов и парциальных пароксизмов.
При неврологическом обследовании у большинства больных (81.8%) были выявлены нарушения. Отмечались: пирамидный гемисиндром (анизорефлексия, анизотония в сочетании с появлением патологических рефлексов, но при отсутствии двигательных расстройств) — 45.5%, координаторные нарушения (неустойчивость в позе Ромберга, легкий интенционный тремор и мимопопадание с одной стороны) — 27.3%. Более, чем у половины пациентов (54.5%) наблюдалось снижение интеллекта в сочетании с гиперактивным поведением.
При проведении исследования ни в одном случаене отмечалось нормальных результатов ЭЭГ. Характерным было замедление основной активности фоновой записи (54.5%), появление генерализованной эпилептической активности (63.6%) и региональных изменений (81.8%). Генерализованная эпилептическая активность была представлена пик-волновыми (45.5%)и полипик-волновыми комплексами (18.2%) с частотой3Гц. Выраженная амплитудная асимметрия комплексов при сохранении их билатерально -синхронного характера констатировалась у 36.4%пациентов. У большинства больных (81.8%)наблюдались региональные изменения биоэлектрической активности. Констатировалось замедление в ритме тета (54.5%), чаще в центрально-париетальных отведениях, а также региональная пик-волновая активность в височных отведениях (27.3%). Проведение гипервентиляционной пробы и ритмической фотостимуляции существенно не изменяло характера ЭЭГ.
КТ и МРТ исследования выявили изменения у 45,5% пациентов. Наблюдались: некрупные арахноидальные кисты по конвекситальной поверхности мозга (18.2%), субатрофия коры головного мозга (9.1%), умеренная вентрикуломегалия (9,1%). У одного пациента при проведении МРТ была выявлена аномалия развития головного мозга с гипоплазией левого полушария и асимметричной вентрикуломегалией.
В лечении МАЭ были апробированы следующие препараты: барбитураты, карбамазепины, сукцинамиды, вальпроаты и ламотриджин. При применении барбитуратов существенное урежение частоты эпилептических приступов было достигнуто у 37.3% больных, однако во всех случаях отмечалось резкое нарастание гиперактивности у детей. Назначение карбамазепинов ни в одном случае не оказало существенного влияния на частоту приступов. У одного пациента с абсансами применение карбамазепина привело к значительному учащению приступов. При монотерапии вальпроатами (депакин, конвулекс, апилепсин) урежение приступов отмечалось в 45.5%случаев. Наиболее оптимистичные результаты были получены при применении производных вальпроевой кислоты в сочетании с другими препаратами. При применении вальпроатов (900 — 2000 мгсут) с ламотриджином (25 — 50 мгсут) удалось достичь полной терапевтической ремиссии у 30.0% больных и значительного улучшения у 50% (из числа принимавших вальпроаты). Комбинация вальпроатов с сукцинамидами или бензодиазепинами вызывала выраженное урежение частоты приступов у большинства больных (60.0%), однако ни в одном случае не была достигнута ремиссия. Таким образом, в общей группе больных МАЭ полная ремиссия была достигнута в 27.3% случаев, значительное урежение приступов — 63.6% и отсутствие эффекта — 9.1% (в том числе у больного с пороком развития головного мозга). Катамнез прослежен в течение 1-3 лет.
Обсуждение
МАЭ относится к редким формам эпилепсии и частота ее, по данным Doose (Rojer), составляет 2% среди всех форм эпилепсии у детей. Согласно результатам собственных исследований, частота МАЭ составляет 5.6% среди генерализованных форм эпилепсии. Таксономическое положение МАЭ в ряду других эпилептических синдромов до конца неясно. С идиопатическими формами эпилепсии МАЭ сближают следующие особенности: преимущественно генерализованный характер приступов(миоклонические, миоклонически-астатические и абсансы), обнаружение на ЭЭГ генерализованной пик- и полипик-волновой активности 3 Гц, положительный терапевтический эффект препаратов вальпроевой кислоты. Ряд особенностей сближает МАЭ с симптоматическими формами эпилепсии: возможность появления парциальных приступов, высокая частота обнаружения очаговой неврологической симптоматики и интеллектуальных нарушений, относительная терапевтическая резистентность. Таким образом, данный синдром является как бы переходным от идиопатических генерализованных форм эпилепсии (например, абсансные формы эпилепсии) к симптоматическим формам (синдромЛеннокса-Гасто).
Ряд авторов подвергает сомнению нозологическую самостоятельность МАЭ (Willye). По мнению Aicardi, МАЭ является особым (так называемым «миоклоническим») вариантом СЛГ. Вместе с тем, точная диагностика двух данных синдромов является принципиально важной в отношении тактики лечения, течения и прогноза заболевания. Нами представлены дифференциально-диагностические критерии МАЭ с СЛГ. Кардинальным отличием клинических проявлений двух данных синдромов является наличие миоклонически-астатических приступов при МАЭ и атонически-астатических при СЛГ. Данное отличие особенно заметно при видео — мониторинге приступов. Как правило, миоклонически-астатические приступы начинаются с коротких миоклонических подергиваний нижних конечностей, после чего происходит падение пациентов на колени или ягодицы. При атонически-астатических приступах возникает внезапное снижение мышечного тонуса и вертикальное падение пациентов с тяжелой травматизацией. На возможность возникновения абсансов при обеих формах эпилепсии указывалось рядом авторов (Guerrini). Подчеркивается различный характер абсансов: типичные (частота пик -волновых комплексов 3 и более Гц) при МАЭ (Beck-Mannagetta) и атипичные (комплексы острая — медленная волна низкой частоты — меньше 2.5 Гц) абсансы — при СЛГ(Zifkin ). отмечает при СЛГ отсутствие прямой связи между возникновением эпилептиформных ЭЭГ паттернов и появлением клинических пароксизмов. Вместе с тем, в большинстве случаев при МАЭ имеется четкая зависимость между появлением пик-и полипик-волновых комплексов и возникновением абсансов и миоклонических подергиваний. Кроме этого, типичным для СЛГ является выраженное замедление основной активности фоновой записи ЭЭГ и появление комплексов острая — медленная волна, как генерализованных, так и мультифокальных. Данные изменения нетипичны для МАЭ. Одним из важных диагностических признаков при СЛГ является возникновение до момента дебюта приступов или сразу после их начала быстрого снижения интеллекта.
Таким образом, проведенное исследование показало клиническое и электронейрофизиологическое своеобразие МАЭ, что свидетельствует, по нашему мнению, о нозологической самостоятельности данного синдрома. Согласно полученным результатам,критериями диагноза МАЭ являются:
дебют приступов в дошкольном возрасте (максимум в 2-3 года),
миоклонические и миоклонически-астатические приступы как основной вид пароксизмов, высокая частота наличия ГСП, с которых нередко дебютирует заболевание, возможность появления парциальных приступов, высокая частота обнаружения неврологических расстройств и снижения интеллекта, относительная резистентность к монотерапии базовыми антиконвульсантами.
Представленные диагностические критериипозволят выделить МАЭ из других миоклонических форм эпилепсии у детей и назначить адекватную антиконвульсантную терапию. Оптимальной терапевтической схемой является сочетание вальпроатов с ламикталом или сукцинамидами или бензодиазепинами.
Библиографический указатель
Zifkin B.G.- «The Lennox — Gastaut syndrome».- In: Comprehensive epileptology, — eds. M.Dam & L.Gram,- 1990,- pp.123-131. New York, Raven Press.
Aicardi J.- «Epilepsy in children».- 1994. New York, Raven Press.
G.Beck-Mannagetta, C.Dehe-Steffens, B.Schmits, D.Jans.- «Genetic aspects of epilepsies of early childhood with mioclonic — astatic seizures».- In: Epileptic seizures and syndromes,- eds. P.Wolf,- 1994, -pp. 165-168. London, John Libbey.
R.Guerrini, Ch.Drave, G.Gobbi et al.- «Idiopathic generalized epilepsies with myoclonus in infancy and childhood».- In: Idiopathic generalized epilepsies,- eds. A.Malafosse, P.Genton, E.Hirsch et al.,- 1994,- pp. 267-280. London, John Libbey.
Wyllie E.-«The treatment of epilepsy».-1993,-1238 P. Philadelphia. Lea & Febiger.
J.Roger et al.- «Epileptic syndromes in infancy, сhildhood, adolescence». — Paris — 1992-p.316-327 (классификация).
H.Jackson.- «On a case of fits resembling those artificially produced in guinea — pigs».- In: Selected writings of John Hughlings Jackson.- 1931.- V.1.- p. 362. London, Hodder & Stought
Клиническое обследование больного с целью постановки диагноза.
Это обследование — достаточно простое. Для него необходим тщательный сбор анамнеза и повторные видео-ЭЭГ записи, чтобы продемонстрировать наличие МП с генерализованными СВ разрядами, спонтанными или такими, которым способствуют сонливость, шум, контакт или ИРС. Записи сна позволяют показать слабую активацию разрядов и фокальных нарушений. Полезно использовать нейровизуализацию для подтверждения отсутствия поражения мозга, но она не является обязательной при наличии типичной симптоматики. Более полезно психоневрологическое обследование для проверки психомоторного развития и прослеживания течения заболевания.
Лечение
ВПА в монотерапии является препаратом выбора, его применение следует начинать как можно раньше. Предпочтительнее применять раствор, а не сироп, так как он лучше переносится ребенком. Необходимо строго следить за концентрацией препарата в плазме.
Обычно бывает достаточно использовать дневную дозу 30 мг/кг 3 раза / день, но для некоторых больных может потребоваться и более высокая доза. ВПА также эффективен при возможных фебрильных припадках. Если ВПА дает неполный эффект, можно использовать его в сочетании с бензоадиазепином (СLВ или НТЗ) или ETS и пересмотреть диагноз. Лечение следует проводить в течение 3-4 лет после начала заболевания, если оно хорошо переносится. В случаях чисто рефлекторных признаков медикаментозную терапию можно не применять. Если она уже начата, ее можно резко прекратить, однако если отсутствует сверхчувствительность. Если БЭП случается у подростка, можно провести кратковременное лечение в этом возрасте. Консервативное лечение должно сочетаться с психологической помощью семье.
Лечение юношеской миоклонической эпилепсии
Режимные мероприятия
Важное значение имеет не только фармакотерапия эпилепсии, но и соблюдение пациентом некоторых жизненных норм, позволяющих избегать провоцирования приступов. Как и при других видах эпилепсии, при ЮМЭ приступы могут быть вызваны нарушением режима, психической и физической перегрузкой, стрессом, недосыпанием, приемом содержащих алкоголь напитков. Поэтому пациенту следует избегать подобных провоцирующих факторов. Положительно влияет на течение заболевания спокойный, простой и неторопливый уклад жизни, пребывание на природе, вдали от городской суеты. В связи с этим некоторые семьи, где у ребенка диагностирована ЮМЭ, переезжают и живут в сельской местности.
Фармакотерапия
Медикаментозная терапия ЮМЭ проводится вальпроатами. Монотерапия данными препаратами оказалась эффективной в отношении всех видов приступов, сопровождающих клинику ЮМЭ, — миоклонических, тонико-клонических и абсансов. При недостаточности монотерапии возможно комбинированное лечение. Купирование резистентных абсансов достигается сочетанием вальпроатов с этосуксимидом, резистентных клонико-тонических приступов — сочетанием вальпроатов с примидоном или фенобарбиталом.
Для контроля миоклонических пароксизмов эффективен клоназепам, однако его действие не распространяется на тонико-клонические генерализованные приступы. При этом полное купирование миоклонических приступов лишает пациента возможности заранее знать о приближающемся тонико-клоническом приступе по возникающим перед ним миоклоническим проявлениям. Поэтому назначение клоназепама оправдано лишь при стойких миоклонических пароксизмах и должно сочетаться с препаратом вальпроевой кислоты.
Результаты лечения ЮМЭ противоэпилептическими препаратами нового поколения (леветирацетамом, ламотриджином, топираматом) пока проходят проверку в клинических условиях. Отмечена высокая перспективность леветирацетама.