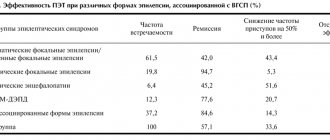
Partial epilepsy is a type of disease that occurs when there is a limited area of activity in the brain. Accompanied by focal or secondary generalized seizures. Accounts for 82% of all variants of epileptoid syndrome. In 70% of cases it is formed against the background of injuries and other pathologies. In 75% of patients, it debuts in childhood.
The specialists at the Leto clinic have extensive practical experience in treating epilepsy. Registration is carried out by telephone.
Causes
Partial seizures of epilepsy occur under the influence of the following factors:
- abnormalities of limited areas of the brain: cysts, arteriovenous malformations, areas of dysplasia in the cortex;
- cerebral infections: cysticercosis, abscesses, encephalitis of various origins, neurosyphilis;
- vascular pathologies: hemorrhages in the medulla;
- TBI;
- neoplasms;
- dysmetabolic encephalopathies.
During development in children, intrauterine infections and complications during pregnancy and childbirth play a significant role: fetal hypoxia, asphyxia during passage through the birth canal. Sometimes symptoms are provoked by disturbances in the maturation of cortical structures and disappear as they grow older.
Generalized seizures
In cases where the foci of the disease are located in both hemispheres of the brain, there is a possibility of developing a generalized attack of epilepsy. Such attacks pose a significant danger to humans, since in the vast majority of cases they end in loss of consciousness. Deaths cannot be ruled out.
According to clinical signs, generalized seizures are divided into:
- Absence - twitching of the eyelids and facial muscles, lack of response to external stimuli, in addition, increased breathing and rolling of the eyes, blackout (10-30 seconds) may be observed.
- Tonic-clonic - a sudden onset of convulsions and complete loss of consciousness (up to 3-5 minutes), holding your breath, urinary incontinence, possibly biting your tongue, foaming at the mouth. After the attack ends, the person usually falls asleep. Amnesia.
- Myoclonic - short-term loss of consciousness (2-10 seconds), jerky twitching of the body.
- Atonic (akinetic) - sudden numbness of a part of the body or loss of tone of one or more muscle groups. Often ends in jaw dropping or falling.
- Tonic - convulsive muscle contractions, flexion and extension of the arms.
Treatment programs
Treatment of depression
Panic attacks
Treatment of schizophrenia
Neuroses, phobias
The lack of appropriate therapy or neglect of the recommendations of the attending physician can lead to a sharp deterioration of the condition and immediate contact with an ambulance. Among the possible complications that threaten the patient's life are the following:
- Status epilepticus is an extremely serious condition in which the patient does not regain consciousness for more than 30 minutes. In this case, immediate resuscitation measures are required.
- A series of epileptic seizures with intervals insufficient to restore normal well-being. The patient usually feels unwell and is depressed. This clinical picture requires immediate contact with a neurologist.
Consultation with an epileptologist
24-hour telephone number to contact the doctor on duty: 8 (831) 266-03-06.
Any request for medical help is a medical confidentiality and cannot be disclosed.
Anonymity guaranteed!
Classification
The following types of partial (focal) epilepsy are distinguished:
Symptomatic. Formed as a result of other diseases. It is possible to determine the causes of development and detect corresponding changes based on neuroimaging data.- Cryptogenic. Presumably caused by another pathology, but the morphological basis is not revealed using imaging techniques.
- Idiopathic. There are no provoking diseases. The causes are disorders of brain maturation, hereditary membrane or channelopathies.
Symptoms
The main manifestation is repeated focal seizures with or without loss of consciousness. Simple paroxysms are sensitive, motor, somatosensory, and autonomic. Sometimes hallucinations (perception of non-existent sounds, tastes, smells, visual phenomena) and pathopsychological symptoms are observed.
With complex partial epilepsy, the onset is the same as with simple epileptic seizures. Automatisms and loss of consciousness are possible. After the episode ends, there is a feeling of confusion. Secondarily generalized seizures begin as simple or complex. Then the excitation covers other parts of the brain with the occurrence of tonic-clonic seizures.
Paroxysms are supplemented by symptoms caused by the main lesion of the central nervous system. Secondary seizures are combined with mental retardation, decreased intelligence, and cognitive decline. The idiopathic form is benign. There are no mental, intellectual or neurological disorders. The clinical picture is determined by the localization of the lesion:
- Temporal. Duration - up to 1 minute. Preceded by an aura. Motor and sensory components are pronounced. Possible loss of consciousness. Oral automatisms are more common in childhood, and automatic gestures are more common in adults. In half of the cases, secondary generalization is detected. When the dominant hemisphere is involved after a complex attack, short-term disorders of speech activity are observed.
- Frontal. There is usually no aura. Short-term serial stereotypies are determined. Complex gestures, leg movements reminiscent of riding a bicycle, agitation, screaming, and aggression are possible. Often partial seizures of epilepsy develop during sleep.
- Occipital. Lasts up to 13 minutes. The most typical are illusions, a decrease in the field of vision, and temporary blindness.
- Parietal. Rarely seen. Characterized by somatosensory seizures followed by transient aphasia. Sometimes only sensitivity disorders are observed.
Partial seizures
Seizures that are caused by the location of the focus of the disease in one part of the brain are called partial. Concomitant symptoms make it possible to accurately determine the location of the affected area. In most cases, this is confirmed by an electroencephalogram.
Partial seizures are differentiated into simple and complex. In a simple partial seizure, the patient remains conscious. His emotions, as a rule, are spontaneous and often inappropriate, and his sensations are unrealistic.
Consultations
Epileptologist
Psychiatrist
Neurologist
Psychotherapist
Paroxysms caused by simple partial seizures include:
- Motor - the patient may experience convulsions, spastic muscle contractions, spontaneous twitching of the limbs, uncontrolled gestures, temporary loss of speech.
- Vegetative - characterized by dilated pupils, pressure fluctuations, pallor of the skin or, on the contrary, their redness. In addition, patients may complain of unusual sensations in the abdomen, an attack of hunger and thirst.
- Sensory - numbness or tingling in the limbs, increasing tinnitus, flashes or spots before the eyes, unusual, often unpleasant, smells and sensations in the mouth.
- Mental - speech impairment, memory loss, inappropriate behavior, panic attacks. Sudden mood swings and hallucinations are possible.
A complex or complex partial seizure is characterized by a significant impairment of consciousness in which the patient is aware of what is happening to him, but is unable to make contact with the people around him. In addition, the patient has a feeling of unreality of what is happening.
Diagnostics
New localized seizures may indicate severe damage to the central nervous system. The diagnostic program includes not only confirmation of the presence and determination of the type of epilepsy, but also a detailed examination to identify the provoking pathology. During the conversation, the doctor establishes the characteristics, frequency of occurrence and duration of paroxysms.
During a neurological examination, in the symptomatic form, changes are determined that indicate the localization of the pathological focus. To make a final diagnosis, the following methods are used:
Electroencephalography. Epi-activity is often recorded not only during seizures, but also in the interictal period. If the indicators are normal, provocative tests are performed. To establish the exact localization of the lesion, subdural corticography is performed.- MRI. Recommended for clarifying the underlying pathology. To increase reliability, the minimum slice thickness is selected. Confirms the presence of focal dysplastic or atrophic areas.
- Other methods. Positron emission tomography sometimes visualizes an area of hypometabolism. SPECT in the same area at the time of paroxysm reveals increased blood supply, the rest of the time - deterioration of blood circulation.
Benign partial epilepsy of childhood
Benign partial epilepsies of childhood include:
- benign partial epilepsy with centrotemporal spikes (rolandic epilepsy);
- benign partial epilepsy with occipital paroxysms;
- benign partial epilepsy with affective symptoms (benign psychomotor epilepsy). The question of whether 2 more forms belong to this group is also discussed:
- atypical benign partial epilepsy:
- benign partial epilepsy with extreme somatosensory evoked potentials.
Benign partial epilepsy with centrotemporal spikes. (Rolandic epilepsy)
The disease was first described by Gastaut in 1952.
Frequency. Rolandic epilepsy is relatively common and accounts for 15% of all epilepsies in children under 15 years of age. The frequency of occurrence, in comparison with absence seizures, is 4-7 times higher. Mostly males are affected.
Genetic data. Hereditary burden of epilepsy can be traced in 17-59% of cases (Blom et al, 1972; Blom, Heijbel, 1982).
Clinical characteristics. Rolandic epilepsy most often manifests at the age of 4-10 years (Blom et al, 1972). The attack usually begins with unilateral sensory sensations (numbness, tingling) in the orofaciomandibular region. In the future, tonic tension in the facial muscles may be noted; clonic or tonic-clonic twitching of the limbs is less common. When the muscles of the larynx and pharynx are involved in the process, speech disturbances are noted (inarticulate sounds, complete inability to pronounce sounds). Attacks often occur with preserved consciousness, however, with the generalization of paroxysms, loss of consciousness is possible (Nayrac, Beaussart, 1958). One of the important features of Rolandic epilepsy is the frequent occurrence of attacks at night, mainly during the phase of falling asleep, or shortly before waking up. Attacks are usually rare. They usually occur at intervals of weeks, months, and sometimes occur once. In only 20% of cases the frequency of attacks is relatively high (Lerman, 1985). Intelligence is usually normal (Lerman, 1985; Dalla Bernardina et al, 1992). A decrease in intelligence and changes in behavior are observed only in isolated cases (Beaumanoir et al, 1974; Lerman, 1985). Behavioral disorders are not caused by the disease itself, but are secondary and associated with parental “overprotection” (Lerman, 1985). The thesis about the absolute absence of pathological changes in the central nervous system in rolandic epilepsy is debatable (Morikawa et al, 1979). Lerman (1985) drew attention to the fact that in 3% of cases with Rolandic epilepsy, hemiparesis is observed. School performance in children with rolandic epilepsy is usually satisfactory, and professional skills are easy.
Data from laboratory and functional studies. EEG is a necessary method to verify the diagnosis. Rolandic epilepsy is characterized by the presence of normal basal activity and spikes of sharp waves localized in the centrotemporal areas. The frequency of spikes and sharp waves increases during sleep. During the slow-wave sleep phase, the formation of bilateral or independent foci of epileptic activity is possible (Blom et al, 1972; Dalla Bernardina et al, 1992), and in some cases, the occurrence of generalized outbreaks of “spike-wave” complexes with a frequency
3-4/sec (Lerman, 1985, 1992). It should be noted that in 30% of cases, EEG patterns typical of rolandic epilepsy are recorded only during sleep (Lerman, 1985). At the same time, the sleep profile in rolandic epilepsy is usually not changed (Nayrac, Beaussart, 1958). Noteworthy is the fact that “rolandic adhesions” are sometimes (1.2-2.4% of cases) observed in healthy people (Cavazutti et al, 1980) and in patients with certain neurological diseases (Degen et al, 1988).
The differential diagnosis should primarily be made with simple and complex focal seizures observed in symptomatic partial epilepsies. In Rolandic epilepsy, in comparison with symptomatic partial epilepsy, intelligence is usually normal, there are no pronounced behavioral disorders, and no pathological changes are noted in neuroradiological studies. Typical patterns are recorded on the EEG - normal basic activity and spikes of centrotemporal localization.
The most difficult differential diagnosis is rolandic epilepsy and complex partial paroxysms combined with impaired consciousness (Lerman, 1985). Difficulties in differential diagnosis are associated with the erroneous interpretation of speech disorders in rolandic epilepsy as a disorder of consciousness, as well as the impossibility of adequately assessing disorders of consciousness. occurring at night. In case of temporal and frontal epilepsy, the EEG reveals focal changes in the corresponding areas, and in case of rolandic epilepsy, spikes of centrotemporal localization are detected.
Treatment. Sulthiam has a satisfactory effect in the treatment of rolandic epilepsy (Doose et al, 1988). The dose of the drug is determined by the clinical manifestations of the disease. A spike-wave is required, as well as rolandic spikes localized in the centrotemporal or parietal areas. In addition, irregular “spike-wave” complexes with a frequency of 3/sec are recorded upon awakening or generalized slow “spike-wave” complexes.
Differential diagnosis should be made with a number of prognostically serious epileptic syndromes of childhood - Lennox-Gastaut syndrome, myoclonic-astatic epilepsy, as well as ESES syndrome. The most common diagnostic error is to consider atypical benign epilepsy as Lennox-Gastaut syndrome. When making a differential diagnosis, it is necessary to take into account that Lennox-Gastaut syndrome is combined with a pronounced delay in neuropsychic development. Typical of Lennox-Gastaut syndrome are nocturnal tonic seizures with characteristic EEG patterns in the form of generalized slow “spike-wave” complexes with a frequency of 2-2.5/sec. An increase in the frequency of these complexes during sleep in Lennox-Gastaut syndrome is an exception.
Myoclonic-astatic epilepsy, in comparison with atypical benign partial epilepsy, is characterized by a decrease in intelligence, there are no partial paroxysms, as well as slow spike-wave complexes during sleep.
Differential diagnosis of atypical benign partial epilepsy and ESES syndrome is based on the results of a polysomnographic study, which reveals typical patterns,
Treatment. The attacks usually disappear spontaneously. Since there is no decrease in intelligence in atypical benign partial epilepsy, combination anticonvulsant therapy is not recommended.
Benign partial epilepsy of childhood with occipital paroxysms
Benign partial epilepsy of childhood with occipital paroxysms was first described by Gastaut in 1950.
Genetic data. The role of genetic factors in the genesis of the disease is undeniable. Many patients have a hereditary history of epilepsy and migraine (Kuzniecky, Rosenblatt, 1987).
Clinical characteristics. The disease manifests itself mainly at the age of 2-8 years. At the same time, an earlier onset of the disease has been described. Gastaut (1992) noted that the disease can manifest itself at 5-17 years of age. The most typical manifestations of seizures in this form of epilepsy are visual disturbances - simple and complex visual hallucinations, illusions, amaurosis (Gastaut, 1992). These symptoms can be isolated or combined with convulsive paroxysms and transient hemiparesis. Often during attacks, headache, vomiting, turning of the head and eyes, and in some cases dysesthesia and dysphagia are noted (Kivity, Lerman, 1992). Attacks with prolonged impairment of consciousness have been described - for 12 hours (Kivity, Lerman, 1992). Panayiotopoulos (1989) noted that most often attacks occur at night and are accompanied by varying degrees of impairment of consciousness, vomiting, and deviation of the eyeballs.
Data from laboratory and functional studies. An EEG study records normal basic activity, high-amplitude (200-300 mV) unilateral or bilateral sharp waves or spike-wave complexes localized in the occipital regions. These changes disappear when you open your eyes. Hyperventilation and photostimulation do not affect the frequency and nature of epileptic EEG patterns. In 38% of cases, EEG studies also reveal generalized bilaterally synchronous “spike-wave” or “polyspike-wave” complexes.
Differential diagnosis should be made with simple and complex partial paroxysms, Lennox-Gastaut syndrome, basilar migraine. Symptomatic partial epilepsies caused by structural damage to the occipital lobe are excluded on the basis of anamnesis, neurological status and neuroradiological examination, which, as a rule, reveal pathological changes. In symptomatic occipital epilepsies, compared with benign occipital epilepsies, opening the eyes does not block epileptic activity on the EEG.
Differential diagnosis with Lennox-Gastaut syndrome is based on the presence of tonic seizures and EEG patterns characteristic of this syndrome. With basilar migraine, there is no epileptic activity on the EEG. Treatment. The first choice drug is carbamazepine.
Benign partial epilepsy with affective symptoms. (Benign psychomotor epilepsy)
Many authors propose to consider benign partial epilepsy with affective symptoms as an independent nosological form (Plouin et al, 1980; Dalla Bernardina et al, 1992).
Clinical characteristics. The disease manifests itself at the age of 2-9 years. The leading symptoms are attacks of fear. These paroxysms occur both during the day and at night. There were no differences in the clinical manifestations of daytime and nighttime paroxysms. The most typical signs of attacks in benign psychomotor epilepsy are paroxysms of fear: the patient suddenly becomes frightened, may cling to the mother, and sometimes swallowing and chewing automatisms occur. Laughter, groans, vegetovisceral disorders (hyperhidrosis, drooling, abdominal pain), and in some cases, speech stoppage (Dalla Bernardina et al, 1992) may also be observed. It should be emphasized that with this form of epilepsy, tonic, tonic-clonic, and atonic seizures are not observed.
Data from laboratory and functional studies. An EEG study reveals normal basic activity, rhythmic spikes or sharp-slow wave complexes with a predominant localization in the frontotemporal or parietotemporal regions (Dalla Bernardina et al, 1992). The frequency of epileptic patterns increases during slow-wave sleep.
Differential diagnosis should be made with complex partial paroxysms, Rolandic epilepsy, and nightmares.
Differential diagnosis of benign partial epilepsy with affective symptoms and complex partial paroxysms is based on the criteria characteristic of this disease: onset in early childhood, normal intelligence, no changes in neurological status and in neuroradiological examination. In some cases, the differential diagnosis with Rolandic epilepsy is quite difficult. As is known, Rolandic epilepsy is often accompanied by speech impairment, guttural sounds, and drooling. A similar symptom complex is sometimes observed in benign partial epilepsy with affective symptoms. Benign partial epilepsy with affective symptoms is supported by pronounced psychomotor symptoms, which are always present in this disease.
Nightmares are very similar in clinical manifestations to seizures observed in benign psychomotor epilepsy. Compared to benign psychomotor epilepsy, nightmares occur exclusively at night, are characterized by frequent seizures and the absence of epileptic patterns on the EEG.
Treatment. Carbamazepine and phenytoin have a satisfactory effect in the treatment of benign psychomotor epilepsy. The prognosis of the disease is favorable.
Benign partial epilepsy with extreme (giant) somatosensory evoked potentials
Dawson in 1947 was the first to show that during somatosensory stimulation in individual patients with partial paroxysms and damage to somatosensory regions, high-amplitude (up to 400 mV) evoked potentials arise. These potentials are recorded on the side contralateral to stimulation. The highest amplitude of potentials is observed in the parasagittal and parietal areas. De Marco in 1971 published the results of a unique observation where touching the outer edge of the foot caused epileptic patterns on the EEG. Later, De Marco and Tassinari (1981) conducted a large study, analyzing 25,000 electroencephalograms from 1,500 children. It has been found that in 1% of children, sensory stimulation of the heels, toes, shoulders, arms or hips provokes giant somatosensory evoked potentials. It is noteworthy that 30% of the children examined suffered from epileptic paroxysms; in 15%, epileptic paroxysms arose later in life.
Clinical characteristics. Giant somatosensory evoked potentials occur between the ages of 4 and 6 years, most often in boys. Patients often have a history of febrile seizures. In most cases, attacks occur during the day. Most often, paroxysms manifest themselves as motor symptoms with a versive component, but generalized tonic-clonic seizures without previous focal disorders are also possible. Epileptic somatosensory potentials persist even after epileptic paroxysm. Attacks are relatively rare, 2-6 times a year. Intelligence does not suffer.
Data from laboratory and functional studies. At the age of 2.5-3.5 years, giant somatosensory evoked potentials appear; later, spontaneously occurring focal epileptic patterns are recorded on the EEG, observed first only during sleep and then during wakefulness. A year after the appearance of epileptic patterns on the EEG, clinically pronounced epileptic paroxysms appear.
Differential diagnosis. The most difficult differential diagnosis is with Rolandic epilepsy. The criteria that support the diagnosis of “benign partial epilepsy with giant somatosensory evoked potentials” are: intact facial muscles at the time of the attack, parasagittal and parietal localization of EEG patterns, spontaneous disappearance of epileptic somatosensory evoked potentials.
Treatment. Anticonvulsant therapy is ineffective. Epileptic seizures usually disappear spontaneously
Landau-Kleffner syndrome
The syndrome was first described by Landau, Kleffner in 1957. To date, about 200 cases of the disease have been described (Deonna, 1991).
Genetic data. According to Beaumanoir (1992), hereditary history of epilepsy is often observed.
Clinical characteristics. The disease manifests itself at the age of 3-7 years (Deonna, 1991). The triad of symptoms is aphasia, epileptic seizures, and behavioral disorders. Early symptoms are progressive speech impairment and verbal agnosia (Pauquier et al, 1992). Speech disorders are characterized by the appearance of speech perseverations, paraphasias, and jargon-aphasia. In most cases, there are no speech dysfunctions preceding the development of the disease (Echnne, 1990; Deonna, 1991). Aphasic disorders can have a fluctuating course with short-term remissions (Deonna et al, 1989). In 2/3-3/4 cases, epileptic paroxysms develop. Seizures are usually simple partial motor seizures. Less common are generalized tonic-clonic, hemiclonic or complex partial seizures and absence seizures. Atonic and tonic paroxysms are extremely rare. In 1/3 of patients, epileptic seizures are rare. Status epilepticus develops extremely rarely. One of the features of epileptic paroxysms in Landau-Kleffner syndrome is their nocturnal nature. The attacks are usually short. Behavioral disorders are manifested by aggression, hyperactivity, and autism.
Data from laboratory and functional studies. An EEG study records normal basic activity, focal or multifocal spikes, sharp waves, spike-wave complexes with predominant localization in the temporal, parieto-temporal or parieto-occipital regions. In some cases, with Landau-Kleffner syndrome, Rolandic spikes are detected on the EEG. A typical EEG pattern in Landau-Kleffner syndrome is electrical status epilepticus during slow-wave sleep (ESES) (Rodriguez and Niedermeyer, 1982).
Neuroradiological examination showed no pathological changes. Spectral positron emission tomography reveals decreased perfusion in the left middle frontal gyrus and right mediotemporal region (Mouridson et al, 1993). Positron emission tomography reveals metabolic disorders in the temporal region (Maquet et al, 1990), which persist even after clinical improvement. Differential diagnosis should, first of all, be carried out with diseases combined with aphasia - tumors, infections, metabolic disorders.
Treatment. Anticonvulsant therapy for speech disorders is practically ineffective (Marescaux et al, 1990). ACTH has a beneficial effect, however, its long-term use is impossible. Significant attention in complex therapy should be paid to speech therapy sessions in order to correct speech disorders.
<< Return to top
Continued >>
Treatment
Involves constant use of anticonvulsants: phenobarbital, levitiracetam, valproic acid derivatives. Etiopathogenetic treatment is aimed at eliminating or mitigating the underlying pathology. Neurometabolites, vascular agents, vitamins, antibacterial and antiviral drugs are prescribed.
Ineffectiveness of conservative therapy and resistance to pharmacological support are indications for surgery. The purpose of the intervention is excision of the pathological focus (tumor, cystic formation) or direct resection of the epi-focus. The effectiveness of surgical methods increases with clearly localized areas of epileptogenic activity.
Cost of services
| CONSULTATIONS OF SPECIALISTS | |
| Initial consultation with a psychiatrist (60 min.) | 6,000 rub. |
| Repeated consultation | 5,000 rub. |
| Consultation with a psychiatrist-narcologist (60 min.) | 5,000 rub. |
| Consultation with a psychologist | 3,500 rub. |
| Consultation with Gromova E.V. (50 minutes) | 12,000 rub. |
| PSYCHOTHERAPY | |
| Psychotherapy (session) | 7,000 rub. |
| Psychotherapy (5 sessions) | 30,000 rub. |
| Psychotherapy (10 sessions) | 60,000 rub. |
| Group psychotherapy (3-7 people) | 3,500 rub. |
| Psychotherapy session with E.V. Gromova (50 minutes) | 12,000 rub. |
| TREATMENT IN A HOSPITAL | |
| Ward for 4 persons | 10,000 rub./day |
| Ward for 3 persons | 13,000 rub./day |
| Ward 1 bed VIP | 23,000 rub./day |
| Individual post | 5,000 rub. |
| PETE | 15,000 rub./day |
This list does not contain all prices for services provided by our clinic. The full price list can be found on the “Prices” , or by calling: 8(969)060-93-93. Initial consultation is FREE!
Forecast
The prognosis depends on the type of disease. Idiopathic epilepsy is characterized by a benign course, maintaining the usual level of mental activity and cognitive abilities. Sometimes it disappears spontaneously after puberty. In the symptomatic form, the outcome is determined by the underlying disease.
In the absence of serious cerebral changes, adequate selection of drug therapy makes it possible to completely eliminate attacks or significantly reduce their frequency. Significant improvement after surgery is observed in 60-70% of patients, complete recovery in 30%.
At the Leto clinic you can get qualified help for partial forms of epilepsy. High-quality diagnostics and competent medicinal support will allow you to solve the problem and return to a full life. Sign up by phone..
Possible complications
Adult patients diagnosed with epilepsy should be under the supervision of a specialist and promptly receive appropriate treatment. In the absence of adequate therapy, brain lesions will grow, which will lead to a deterioration in the general condition and a reduction in the intervals between attacks.
Any patient diagnosed with epilepsy should be clearly aware that an irresponsible attitude towards one’s own health increases the risk of death from a fall or asphyxia during an attack.
It is especially important to monitor the condition of expectant mothers who have a history of epilepsy. This is due to the fact that an epileptic seizure can lead to fetal death.
Despite the insufficient knowledge of the nature of epilepsy, modern epileptology has significantly expanded the understanding of methods of its treatment and prevention. To get advice from leading specialists in Nizhny Novgorod and start treatment on time, contact the Family Practice clinic.