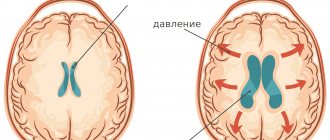
Angelman syndrome is a genetic abnormality. It is characterized by mental retardation, sleep disturbances, seizures, chaotic movements (especially of the hands), frequent laughter or smiling. This disease is also called “Parsley syndrome” or “happy doll” syndrome.
In Angelman syndrome, some genes from the long arm of chromosome 15 are missing (in most cases, a partial deletion or other mutation of chromosome 15). In Angelman syndrome, the maternal chromosome is affected; If the paternal chromosome is damaged, Prader-Willi syndrome occurs.
Treatment
Angelman syndrome is an incurable genetic abnormality. Currently, medical scientists are actively developing effective therapeutic techniques. If the syndrome was diagnosed during fetal development, experts recommend terminating the pregnancy. A newborn sick child requires careful care, care and highly qualified therapy.
Symptomatic treatment helps alleviate the condition of patients with marionette syndrome. They are prescribed medications and non-drug procedures:
Anticonvulsants reduce the frequency and severity of seizures. Patients are prescribed several anticonvulsants at the same time, since attacks are characterized by several types of seizures. The most popular drugs include: Valproic acid, Convulex, Lamotrigine, Clonazepam. These drugs prevent seizures, improve the mood and mental state of patients. Vitamin therapy is the intake of vitamins B, C, D and E. Treatment with vitamins should only be carried out as prescribed by a doctor, since they reduce the effectiveness of antiepileptic drugs. Sleeping pills that improve sleep in easily excitable patients - Melatonin, Diphenhydramine. When problems with digestion and stool occur, patients are prescribed laxatives – “Senade”, “Slabilen”, “Fitolax”, pre- and probiotics – “Hilak Forte”, “Linex”, “Bifiform”. Hormonal therapy is indicated in extreme cases when it is impossible to correct the behavior of patients in other ways.
They are injected with the hormone secretin, which normalizes digestion and has a positive effect on patients’ attention, as well as oxytocin, which improves the child’s cognitive abilities, memory, and behavior. A similar hormone treatment regimen was developed by American scientists for the treatment of children with autism. For Angelman syndrome, behavioral therapy, work with a psychologist, speech pathologist and speech therapist are indicated. Physiotherapeutic procedures help cope with muscle hypotonia and joint problems
Doctors usually prescribe paraffin baths, electrophoresis, and magnetic therapy. Exercise therapy, professional massage, and aqua gymnastics in cool water help patients stand more confidently on their feet. To get rid of hypersalivation, drugs are used that inhibit salivation and surgical interventions aimed at reimplantation of the salivary ducts. Traditional medicine, like homeopathic medicines, are weakly effective in treating Angelman syndrome, but are absolutely safe. A collection based on peony, licorice and duckweed reduces the frequency of convulsive attacks, a decoction of lavender and an aqueous infusion of motherwort have a general calming effect.
Abnormalities of the AZF locus
There is also a special region on the Y chromosome that controls the process of sperm production. This is what determines how effective spermatogenesis will be. In addition, the condition of this area affects the properties of sperm, such as the total number in the ejaculate, the ability to move, the presence of structural changes and the ability to fertilize. Only in the presence of well-formed motile sperm can male genetic material be delivered to the egg. In other words, a man’s ability to have children depends on the state of this small section of genetic code.
If there are abnormalities in the AZF locus, the process of sperm production is disrupted. As a result, azoospermia and oligozoospermia may develop. With these pathologies, the ejaculate either does not contain sperm at all, or their number is greatly reduced.
The AZF locus itself is divided into three parts with specific tasks. They are named by adding a suffix: AZFa, AZFb and AZFc. The resulting deletion can remove a fragment of a separate part, or its entirety, or capture two regions at once. With complete removal of the AZF, severe damage to spermatogenesis develops. Partial deletions can manifest themselves in different ways. At the same time, the degree of manifestation of pathology is influenced by the size of the lost fragment and its location in the locus. Therefore, for prognostic purposes, it is extremely important to know where the deletion occurred. In addition, this information can be used for proper family planning and in vitro fertilization.
If the deletion removed the entire locus or any of the regions with a/b indices, then the man cannot produce viable sperm. If the deletion can be described by the formula AZFb/AZFb+, then azoospermia develops due to severe disturbances in the process of sperm formation.
Deletions of the AZFc region lead to the manifestation of pathological symptoms of varying severity. It is also possible to develop oligospermia, which in principle allows conception. In 50-70 percent of the total number of such cases, it is possible to obtain sperm for further use in artificial insemination methods. Partial deletion of the AZFc region can be expressed in the form of various disorders from normozoospermia to azoospermia.
All deletions in the AZF locus that cause one or another pathological situation are the causes of male infertility. Determination of the mutation is possible by histological analysis of seminal fluid. In this case, it is necessary to stop sperm maturation or detect immature sperm. To obtain accurate data on deletions in the AZF locus, PCR of 6 markers is used, which relate to individual sections of the locus.
Causes
Angelman syndrome is associated with mental and neurological developmental delays
It is impossible to identify unambiguous factors for the occurrence of pathology in children. In most cases, a change in the composition of chromosome 15 is determined, but the defects in it are of a different nature. In 10% of patients, chromosomal abnormalities are not detected.
Possible changes in chromosome 15:
- deletion, i.e. loss of part of the genetic material. Angelman syndrome is often diagnosed with a 15q12 deletion. This area is associated with the activation of a large number of genes involved in the development of the nervous system and internal organs;
- in 3–5% of pathological cases, geneticists detect uniparental disomy. This is a condition in which the child’s karyotype contains two paternal chromosomes 15. There is no maternal copy;
- In the prenatal period of development, the UBE3A locus, localized on chromosome 15, plays an important role. When it is epigenetically “switched off,” symptoms of the disease develop;
- a mutation in the UBE3A locus is detected in 5–10% of sick children.
Patients with pathology and their parents need consultation with a geneticist. The specialist conducts molecular genetic studies aimed at identifying the cause of Angelman syndrome.
Forms
Angelman syndrome is associated with four types of genetic mutations:
- A newly emerged chromosomal mutation, which is associated with the loss of a section of the chromosome at locus 15 q11 - q13. This mutation is the cause of about 80% of all cases of the disease.
- Unipaternal disomy, which is associated with the loss of the maternal locus (lack of maternal genetic material). This option is rare (about 5% of all cases).
- A defect in a number of genes subject to genomic imprinting (GI). These defects occur in 2-4% of patients as a result of a direct violation of imprinting (differences in the conversion of gene information into protein or RNA, which depend on the origin of the gene). Most often occurs as a result of loss of the GI regulatory center. GI defects without loss of the regulatory center are the result of a spontaneous mutation, the repetition of which is very rare.
- A spontaneous mutation in the maternal copy that causes the brain copy of the UBE3A gene to fail to transform. This gene encodes the activity of ubiquitin ligase (an enzyme involved in the complex process of protein breakdown). Deficiency of this enzyme is one of the molecular mechanisms of the syndrome.
It is currently not possible to establish the form of the disease in 7-9%.
Angelman syndrome - what is it?
This pathology is a congenital disease. It develops in utero. This disorder was first described by a British pediatrician, Dr. Harry Angelman, in 1965. Back then, patients with this diagnosis were called “doll children.” The disease itself was called “happy puppet syndrome.” This disease has other names. One of them is Parsley syndrome. This pathology is accompanied by delayed physical and mental development, walking on straight legs and laughing for no reason. Outwardly, patients with this diagnosis look like dolls.
Angelman syndrome - causes
To this day, scientists are studying the factors that provoke the development of this pathology. Angelman syndrome develops more often, if genetics are disturbed. In other words, the likelihood of its occurrence increases when one of the parents has chromosomal defects. However, Angelman syndrome, Parsley syndrome, and the laughing doll syndrome can also appear spontaneously in a baby whose parents are completely healthy.
The following factors have a teratogenic effect on a child:
- bad habits of a pregnant woman;
- long-term use of psychotropic drugs by parents;
- strong feelings of the expectant mother, provoked by grief or excessive stress;
- an inflammatory process that affects the reproductive system of a pregnant woman;
- stay of the expectant mother in a zone of increased radiation radiation.
Angelman syndrome - type of inheritance
This pathology occurs due to a disturbance in the process of chromosome division. Angelman syndrome has a karyotype of 46 XX or XY. Defects are found in the 15th chromosome. The mutation manifests itself as follows:
- Deletion
is the loss of a piece of chromosome. Patients with this disorder suffer from severe mental retardation. These children in most cases cannot walk or talk. In addition, such patients are often overcome by epileptic seizures. - Duplication
is the appearance of extra genes. If Angelman syndrome is diagnosed, patients are more likely to die in infancy. Sometimes such people survive until puberty. - Inversion
is a defect in which part of a chromosome is missing. In this case, the genes are located in the reverse sequence. - Translocation
- part of a chromosome is attached to another. - Uniparental disomy
is a disorder in which a child inherits two copies of the father's set of chromosome 15. He does not receive maternal benefits.
Etiology of the disease
This syndrome is quite rare, occurring in approximately 1 case per 10-20 thousand newborns. The main cause of the disease is the loss of a copy of the mother's normal genes on chromosome 15, which occurs due to a disruption in the division of this chromosome. In addition, the disease can occur due to a gene mutation on the paternal side, paternal trisomy or disomy.
Normally, in a healthy person, one copy of chromosome 15 is transmitted from the mother and from the father. If a child receives a slightly genetically altered copy from one of the parents (especially if it is the mother’s gene, since they are stronger than the father’s genes), then the baby will develop Angelman syndrome.
Classification and symptoms of pathology
The clinical picture of the lesion is largely determined by the type of genetic defect that causes its occurrence. In medicine, it is customary to distinguish several types of disease:
- Loss of a section of chromosome 15 is the most common form of karyotype leading to symptoms in Angelman syndrome. It is diagnosed in 80% of cases.
- In 5% of patients, there is a loss of part of the maternal genetic information. A section of chromosome 15, which should be obtained from a woman's egg, is replaced with material from the sperm's DNA. This phenomenon is known as paternal disomy.
- Several genes can mutate at once. This problem is registered only in 3–4% of cases. Angelman syndrome in children with a similar defect is caused by disturbances in the imprinting process. This occurs as a result of mutation of the genes that control this mechanism.
- Some patients experience spontaneous formation of the defect. In such cases, a gene located in the child’s brain is transformed. This structure is responsible for the fermentation of proteins. Symptoms develop against the background of a deficiency of this compound.
In 6–8% of cases, it is not possible to determine the type of lesion.
Angelman syndrome is associated with various clinical manifestations, including:
- Retarded mental and physical development. A specific symptom is a decrease in the volume of the skull. A child with Parsley syndrome is difficult to learn, since the disease is associated with disruption of the brain.
- A characteristic sign of defeat is that the baby tends to constantly smile and laugh. However, there are often no reasons for such behavior. Patients appear cheerful and willing to make contact with other children and adults. It is this manner that allows doctors to suspect the presence of a disorder.
- Neurological symptoms are also common. They include tremor, hyperactivity of the upper limb girdle, and a specific gait. Children move in such a way that their legs remain straight. This gave the original name to the pathological condition - happy doll or puppet syndrome. Some patients experience seizures, which worsen their prognosis.
- Language dysfunction is a common problem in children with Angelman syndrome. At the same time, doctors often note a good understanding of the words of others, combined with difficulty expressing their own thoughts. The problem leaves a serious imprint on the social life of patients.
- Deformations of the skull, its cerebral and facial parts are also possible. Children have a massive chin pushed forward, a wide mouth, as well as anomalies in the formation and development of teeth.
It is characteristic that symptoms intensify on average at the age of 5–7 years. In newborns, the clinical picture is in most cases nonspecific and does not allow one to suspect the syndrome. Specific disorders that confirm the diagnosis are noted in the preschool period and are often associated with a lag behind peers in mental and physical development.
The main problem with the disease remains a decrease in communication skills, as well as difficulties in self-care. About 80% of patients with Angelman syndrome express their thoughts in 3–4 words. Children and adults with a genetic abnormality communicate well using gestures, so they widely use them in everyday life. This characteristic only proves relatively high intellectual abilities. Formal languages remain difficult to access, as a result of which the methods of interaction with other people are individual in each case. It has been proven that patients with Engelman syndrome are unable to think abstractly.
Self-care skills also vary. The vast majority of people with a genetic abnormality are able to walk independently, relieve themselves, and use cutlery. Patients learn to communicate food preferences. They may need assistance with bathing and wearing clothes with buttons and zippers. At the same time, many actions performed by patients in everyday life require control. This need is due to the fact that people with Angelman syndrome completely lack a sense of danger. However, with proper support from loved ones, they adapt well to external conditions.
Anomalies of genetic material
Hereditary material consists of a huge number of nucleotides that form genes. Moreover, in each gene the nucleotide sequence is strictly defined, since it must code for a specific protein. In addition, the genes themselves, when forming chromosomes, are also arranged in a fixed order. Thanks to the preservation of this order, the body can function, and scientists can quickly and accurately indicate to each other which gene we are talking about.
Ideally, the system works without the slightest glitch, and genetic information is always transmitted unchanged. However, in practice, a large number of structural units and constant exposure to various factors (for example, ionizing radiation) leads to the fact that various anomalies arise from time to time. In particular, individual sections of the DNA sequence can be copied to a new location. In this case, they talk about duplication. If, instead of creating a new copy, part of the original chain was moved, then the modification is called translocation. In addition, sometimes part of the sequence is simply lost, removed from the genetic material. In this case, the change is called a deletion.
Since the interactions in the body have been honed over many millennia of evolutionary development, the result is a very coherent system. And anomalies, even the smallest ones, can cause an imbalance. In this case, one or another disorder develops in the body. If the cause is at the level of genes, then they speak of gene diseases. If an extra copy of a chromosome is lost or, on the contrary, gained, such disorders are called chromosomal diseases.
Notes
- X-linked mental retardation. (Russian). Center for Molecular Genetics at the Medical Genetic Research Center of the Russian Academy of Medical Sciences. Retrieved May 8, 2021.
- Yamasaki K., Joh K., Ohta T., Masuzaki H., Ishimaru T., Mukai T., Niikawa N., Ogawa M., Wagstaff J., Kishino T.
Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. (English) // Human molecular genetics. - 2003. - Vol. 12, no. 8. - P. 837-847. — DOI:10.1093/hmg/ddg106. - PMID 12668607. - Petersen MB, Brøndum-Nielsen K., Hansen LK, Wulff K.
Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: estimated prevalence rate in a Danish county. (English) // American journal of medical genetics. - 1995. - Vol. 60, no. 3. - P. 261-262. — DOI:10.1002/ajmg.1320600317. - PMID 7573182. - Steffenburg S., Gillberg CL, Steffenburg U., Kyllerman M.
Autism in Angelman syndrome: a population-based study. (English) // Pediatric neurology. - 1996. - Vol. 14, no. 2. - P. 131-136. — DOI:10.1016/0887-8994(96)00011-2. - PMID 8703225.
Treatment
Angelman syndrome is caused by a congenital genetic abnormality; Currently, specific methods for its treatment have not been developed, but some therapeutic measures improve the quality of life of people with the syndrome. Infants with hypotonia should receive massage and other special therapies (physical therapy).
It is recommended to use special methods of child development, classes with a speech therapist and a speech pathologist. Sleep disturbances are corrected by prescribing mild sleeping pills. Dr. Wagstaff (USA) believes that administering 0.3 mg melatonin 0.5 to 1.0 hours before bed improves sleep in patients with Angelman syndrome. Stool disorders are regulated by the administration of mild laxatives.
For seizures, the treatment approach is the same as for epilepsy. Children with Angelman syndrome often have more than one type of seizure. Electroencephalography is indicated.
Undesirable behavior requires correction. Dr. Charles Williams (Gainesville, Florida), who works primarily with autistic children, notes common behavior patterns among autistic children and children with Angelman syndrome: marked self-stimulation, impulsivity, compulsive, repetitive movements, interest in inappropriate objects, and difficulty in communicating with other people. US doctors report that in autistic children, intravenous injections of the hormone secretin (found in the pancreas) successfully reduce unwanted behavior and promote good levels of sociability and communication skills; Perhaps medicine will come to the use of secretin to correct the behavior of children with Angelman syndrome.
Giovanni Francesco Caroto (c. –). Portrait of a red-haired boy with a drawing of a jointed doll
Forecast
The prognosis for Angelman syndrome depends on the nature of the chromosomal abnormality and the timeliness of its detection. The hardest time is for those children whose 15th chromosome contains “missing” genes (deletions). The likelihood of walking and talking in such patients is extremely low. Other cases can be corrected with an attentive approach and love for your child.
Such patients, alas, will not be able to become full-fledged members of society, despite the fact that they are far from stupid and understand speech and its meaning. It’s just that they have problems with communication for the rest of their lives. Patients can be taught sign language from childhood, but cannot be forced to communicate using words. The vocabulary of “talking” patients is limited to a minimum of words used in everyday life (5-15 words).
As for the life expectancy and general health of patients with Angelman syndrome, the figures here fluctuate around the average. In adulthood, patients mainly face health problems such as scoliosis and obesity, which, if treated correctly, are not life-threatening.
(
2 ratings, average: 5.00 out of 5)
Angelman syndrome causes
In this disease, certain genes on chromosome 15 are missing. Most cases are characterized by a partial deletion or mutation of chromosome 15. With Angelman syndrome, the maternal chromosome is affected, and in case of changes in the paternal chromosome, Prader-Willi syndrome develops. Often the syndrome is caused by a spontaneous chromosomal defect, which is characterized by the absence of a contiguous region consisting of four million base pairs of DNA in places q11-q13 of the 15th chromosome.
Angelman syndrome has a karyotype of 46 XX or XY, 15r. Independent studies identify mutations in the UBE3A gene as the causes of Angelman syndrome. The enzymatic component of a complex protein degradation system is considered the product of this gene.
Diagnostics
Most often, the diagnosis is made when the child reaches the age of 3-7 years. During this period, symptoms of the disease appear, which are used to preliminarily determine its presence.
During the neonatal period, it is quite difficult to identify pathology, since there are no clinical manifestations and are hidden. In addition to analyzing the signs of the disease.
A number of studies are carried out, such as determining the tone of muscle tissue, as well as genetic research on chromosome 15. Violations of this element indicate the presence of Angelman syndrome.
How is Angelman syndrome treated?
This is a genetic disease and, unfortunately, scientists have not yet found methods to treat it. But there are techniques that make life easier for such people and significantly improve their quality of life.
In each individual case, with a disease such as Angelman syndrome in children, the symptoms will be different. Therefore, an individual therapy method is developed for each patient.
There are several types of therapeutic treatment, they are as follows.
- Since Angelman syndrome is characterized by seizures, convulsions, and epileptic attacks, antiepileptic drugs, or anticonvulsants, are recommended. They will significantly reduce the manifestations of these symptoms and allow you to control the patient’s condition.
- Impaired motor skills and problems with motor functions are also symptoms of Angelman syndrome. Physical therapy classes and special training designed for such patients are necessary for their development. You need to do the exercises regularly and patiently, and then the work will be rewarded. Such children develop slowly, but with the patience of their parents a lot can be achieved.
Special classes help children with Angelman syndrome adapt to the outside world
Children with hypotension should be prescribed massage and special physiotherapeutic procedures. If a child has Angelman syndrome, treatment must include sign language. This will be very useful, both for them in further communication with people, and for the mothers and fathers themselves. Classes with a speech pathologist and speech therapist are recommended. Specially designed programs help to correctly and effectively teach children the rules of etiquette and behavior in society
They are designed to help patients cope with increased activity and learn to concentrate their attention. After undergoing such therapy, patients will find it easier to communicate with peers, and some can even start their own families. It is necessary to take a more careful approach to the preparation of the diet, since obesity is very often observed in such children, especially in girls.
In the presence of Angelman syndrome, metabolic processes in the body are disrupted, which can often lead to excess body weight with normal nutrition
- In such patients, as a rule, the stool is disturbed; in this case, laxatives are prescribed.
- For sleep disturbances, mild sleeping pills are prescribed.
Scientists have noticed that Angelman syndrome is very similar to a disease such as autism.
Clinical picture in detail
Different patients may exhibit different symptoms. This difference depends on the degree and type of chromosomal abnormality.
Signs can be divided according to the frequency of their manifestation in children with Angelman syndrome:
- Symptoms that appear in all patients . These include severe functional developmental delay, behavioral deviations (laughter and smiling for no reason, a state of happiness, increased excitability, decreased concentration). Also, absolutely all patients experience impaired motor functions, impaired balance, tremors, a predominance of nonverbal skills over verbal ones, and speech disorders.
- Symptoms characteristic of 80% of patients . Stunted growth leading to disproportion of the head relative to other parts of the body. This often leads to the development of microcephaly. Children under three years of age often experience epileptic seizures, then they become less frequent or disappear altogether. Electroencephalogram results are often abnormal, with low-level waves having increased amplitude and temporal dynamics.
- Symptoms that occur in less than 80% of patients . Such manifestations include strabismus, decreased ability to control tongue movements, problems with swallowing, hypopigmentation of the eyes and skin, albinism. Many patients experience increased activity of tendon reflexes. In early childhood, many people have problems with eating and sleeping. Manifestations also include drooling, tongue protruding, and constant thirst. External signs also include the presence of a flat back of the head and smooth palms.
As you grow older, the manifestations of the syndrome change. Sleep disorders, hyperactivity, and seizures are less common. Adults with Angelman syndrome appear younger than their peers.
Puberty occurs a little later than in individuals who do not have this pathology. Patients can have their own children, but there is a high risk of transmitting the disease to their offspring.
Many adult patients suffer from uncontrollable urination. Often there are difficulties with motor skills, which leads to the need to wear clothes without zippers and buttons. Among adult patients, there is a problem of excess weight, so it is necessary to monitor compliance with a special diet.
Children perceive oral speech well and understand the content of almost all conversations, but rarely respond. They often refuse to take part in the conversation and use several dozen words in their speech.
Symptoms of Angelman syndrome
Manifestations of the disorder in the psycho-emotional sphere:
- developmental delay, including absence of babbling and crawling between 6 months and 1 year;
- learning disability;
- lack of speech or minimal speaking skills;
- unmotivated frequent laughter/smiling;
- the personality type can be described as joyful, exalted.
Symptoms of the disorder at the somatic level:
- seizures of epilepsy, which usually occur between the ages of 2 and 3 years;
- difficulty walking/moving, stiff or sharp angular movements due to the inability to control voluntary movements (ataxia);
- unusual behavior such as sudden flapping of arms, walking with raised arms and/or stiff legs;
- small head size with flattening in the occipital area (microbrachycephaly);
- hair, skin and eyes of a light shade (hypopigmentation);
- Sometimes children have a wide mouth, sparse teeth and a protruding tongue. In most cases, patients' facial features do not differ in anything unusual.
Separately, we should dwell on the features of speech development and motor skills characteristic of people with Angelman syndrome.
Most children with this disorder have an extremely poor vocabulary, which may be limited to a few words (up to one or two dozen). This causes significant difficulties in the socialization of a child who has Parsley syndrome.
At the same time, children with such a disorder - as a rule - understand not only simple commands, but are also able to absorb information in a volume that significantly exceeds their ability to communicate and express thoughts.
As for motor skills, in addition to the manifestations described above, “Parsley” children can often hold their arms raised and bent at the elbows and wrists, and periodically wave their arms in moments of excitement or while walking. It was this similarity to the movements of a puppet, coupled with their angularity and stiffness against the background of the patients’ frequent exaltation, that became the reason for calling the disorder happy doll syndrome.
They may experience both decreased muscle tone in the trunk (hypotonia) and increased muscle tone in the extremities (hypertension), as well as abnormally exaggerated reflex reactions (hyperreflexia). Some children develop fine tremors in the arms and legs. These disorders may appear between 6 months and 1 year of age.
The acquisition of some milestone motor skills, such as walking, is delayed.
In cases of relatively mild forms of the disease, children can begin to walk at 2-3 years of age. In more severe cases, upright walking is slowed down and occurs jerkily - “rigidly”, which also resembles the movements of a puppet. Some children do not walk until they are 5 to 10 years old. Approximately 10% of patients are unable to move without assistance.
The effect of deletions on the ability to fertilize
Deletions that occur on normal chromosomes (autosomes) can in some cases be compensated for by a normal copy of the gene. However, when it comes to sex chromosomes, especially the Y chromosome, the situation changes.
First of all, it should be noted that the genes localized on it do not have a second copy. With a normal number of chromosomes in the set, the Y chromosome turns out to be extremely vulnerable. Combined with a small number of genes, this leads to serious consequences for each change. Of particular interest are mutations affecting the AZF locus and the SRY gene.
Treatment
There is no modern means of preventing and getting rid of parsley (doll) syndrome. There is no treatment for Angelman syndrome today due to the fact that the causes of the genetic defect are unknown. A set of measures has been developed aimed at stabilizing and improving the quality of life in this disease. The rehabilitation program is developed individually, but some of the measures are common to all patients:
- Taking anticonvulsants and antiepileptic drugs.
- Therapeutic preventive physical education, massage, physiotherapy. Helps stabilize general motor skills, improve the functioning of the musculoskeletal system, and teach walking naturally (pictured).
- Learning sign language. Many patients with parsley syndrome use a less than average range of words in conversation, but do not have learning difficulties. Sign language becomes an acceptable alternative in communication if taught from early childhood (pictured).
- Behavior therapy. This method is aimed at compensating for attention deficit, eliminating or suppressing hyperactivity, and providing the opportunity to provide correct education for adaptation in society.
Prevention
It is impossible to prevent the birth of a child with Angelman syndrome, since gene mutation occurs spontaneously. Even the most qualified geneticists cannot explain such a process.
Preventive measures are aimed at thorough preparation for pregnancy and childbirth. All pregnant women must undergo a fetal ultrasound at the appointed time and donate blood for serum markers of chromosomal pathologies. In case of a positive result of a medical genetic study, parents must decide whether they can raise a sick child.
Medical genetic counseling is required for couples with a family history. If this disease has not been recorded in loved ones, it is enough to follow the basic principles of maintaining a healthy lifestyle during pregnancy.
Angelman syndrome is a severe and incurable disease that deprives a person of a normal life. Sick children need special care and attention. Treatment is aimed at reducing the intensity of the symptoms of the pathology. Modern scientists are trying to find effective ways to combat chromosomal abnormalities that affect newborn children.