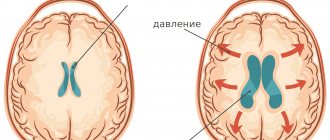
Синдром Ангельмана — генетическая аномалия. Для неё характерны задержка психического развития, нарушения сна, припадки, хаотические движения (особенно рук), частый смех или улыбки. Также эту болезнь называют «синдром Петрушки» или синдромом «счастливой куклы».
При синдроме Ангельмана отсутствуют некоторые гены из длинного плеча 15-й хромосомы (в большинстве случаев — частичная делеция либо другая мутация 15 хромосомы). При синдроме Ангельмана страдает материнская хромосома; в случае повреждения отцовской хромосомы возникает синдром Прадера-Вилли.
Лечение
Синдром Ангельмана — неизлечимая генетическая аномалия. В настоящее время ученые-медики активно разрабатывают эффективные терапевтические методики. Если синдром был диагностирован во время внутриутробного развития, специалисты рекомендуют прервать беременность. Новорожденному больному ребенку необходим тщательный уход, забота и высококвалифицированная терапия.
Симптоматическое лечение помогает облегчить состояние больных с синдромом марионетки. Им назначают лекарственные препараты и немедикаментозные процедуры:
Антиконвульсанты снижают частоту и силу эпиприступов. Больным назначают одновременно несколько противосудорожных средств, поскольку приступам характерны несколько видов судорог. К наиболее популярным препаратам относятся: «Вальпроевая кислота», «Конвулекс», «Ламотриджин», «Клоназепам». Эти средства предупреждают судороги, улучшают настроение и психическое состояние больных. Витаминотерапия — прием витаминов группы В, С, Д и Е. Лечение витаминами следует проводить только по назначению врача, поскольку они снижают эффективность противоэпилептических препаратов. Снотворные препараты, улучшающие сон у легковозбудимых пациентов – «Мелатонин», «Дифенгидрамин». При появлении проблем с пищеварением и стулом больным назначают слабительные средства – «Сенаде», «Слабилен», «Фитолакс», пре- и пробиотики – «Хилак форте», «Линекс», «Бифиформ». Гормональная терапия показана в крайних случаях, когда невозможно другими способами скорректировать поведение больных
Им вводят гормон секретин, нормализующий пищеварение, положительно влияющий на внимание пациентов, а также окситоцин, улучшающий познавательные способности ребенка, память, поведение. Подобная схема лечения гормонами разработана американскими учеными для лечения детей, больных аутизмом. При синдроме Ангельмана показана поведенческая терапия, работа с психологом, дефектологом и логопедом. Физиотерапевтические процедуры помогают справиться с гипотонией мышц и проблемами с суставами
Обычно врачи назначают парафиновые аппликации, электрофорез, магнитотерапию. Увереннее стоять на ногах больным помогает ЛФК, профессиональный массаж, аквагимнастика в прохладной воде. Чтобы избавиться от гиперсаливации, применяют препараты, угнетающие слюнообразование и оперативные вмешательства, направленные на реимплантацию слюнных протоков. Средства народной медицины, как и гомеопатические препараты, слабоэффективны в лечении синдрома Ангельмана, но абсолютно безопасны. Сбор на основе пиона, солодки и ряски снижает частоту судорожных приступов, отвар лаванды и водный настой пустырника оказывают общее успокаивающее действие.
Аномалии AZF-локуса
На Y-хромосоме также имеется особый участок, который контролирует процесс выработки сперматозоидов. Именно от этого зависит, насколько эффективным будет сперматогенез. Кроме того, состояние этого участка сказывается на свойствах сперматозоидов, таких как общее количество в эякуляте, способность двигаться, наличие структурных изменений и способность к оплодотворению. Только при наличии хорошо сформированных подвижных сперматозоидов мужской генетический материал может быть доставлен до яйцеклетки. Иными словами, о состоянии этого небольшого участка генетического кода зависит способность мужчины иметь детей.
При наличии в AZF-локусе аномалий процесс выработки сперматозоидов нарушается. В результате могут развиться азооспермия и олигозооспермия. При этих патологиях в эякуляте либо совсем не содержится сперматозоидов, либо их число сильно снижено.
Сам AZF локус делится на три части со специфическими задачами. Они именуются путем добавления индекса: AZFa, AZFb и AZFc. Возникшая делеция может удалять фрагмент отдельной части, либо ее целиком, либо захватывать сразу два региона. При полном удалении AZF развивается тяжелое поражение сперматогенеза. Частичные делеции могут проявляться по-разному. При этом на степень проявления патологии влияют размеры утраченного фрагмента и его расположение в локусе. Поэтому для прогностических целей крайне важно знать, в каком месте произошла делеция. Кроме того, эта информация может использоваться для правильного планирования семьи и проведения экстракорпорального оплодотворения.
Если при делеции был удален весь локус или любой из регионов с индексами a/b, то у мужчины не могут быть получены жизнеспособные сперматозоиды. Если делецию можно описать формулой AZFb/AZFb+, то развивается азооспермия из-за тяжелых нарушений процесса формирования сперматозоидов.
Делеции участка AZFc приводят к проявлению патологических симптомов различной степени тяжести. В том числе возможно развитие олигоспермии, которая в принципе допускает зачатие. В 50-70 процентах от общего числа подобных случаев возможно получение сперматозоидов для дальнейшего использования в методах искусственного оплодотворения. Частичная делеция региона AZFс может выражаться в форме различных нарушений от нормозооспермии до азооспермии.
Все делеции в AZF-локусе, вызывающие ту или иную патологическую ситуацию, являются причинами мужского бесплодия. Определение мутации возможно путем гистологического анализа семенной жидкости. При этом необходима остановка созревания сперматозоидов или обнаружение незрелых сперматозоидов. Для получения точных данных о делециях в AZF-локусе используется ПЦР 6 маркеров, которые относятся к отдельным участкам локуса.
Причины возникновения
Синдром Ангельмана связан с задержкой психического и неврологического развития
Выявить однозначные факторы возникновения патологии у детей невозможно. В большинстве случаев определяется изменение состава 15 хромосомы, но дефекты в ней носят различный характер. У 10% пациентов хромосомные аномалии не выявляются.
Возможные изменения в 15 хромосоме:
- делеция, т.е. потеря части генетического материала. При синдроме Ангельмана часто определяют делецию 15q12. Этот участок связан с активацией большого числа генов, принимающих участие в развитии нервной системы и внутренних органов;
- в 3–5% случаев патологии генетики обнаруживают однородительскую дисомию. Это состояние, при котором в кариотипе ребенка имеются две отцовских 15 хромосомы. Материнская копия при этом отсутствует;
- во внутриутробном периоде развития важную роль играет локус UBE3A, локализованный на 15 хромосоме. При его эпигенетическом «выключении» развиваются симптомы заболевания;
- мутация в локусе UBE3A выявляется у 5–10% больных детей.
Больные с патологией и их родители нуждаются в консультации врача-генетика. Специалист проводит молекулярно-генетические исследования, направленные на выявление причины появления синдрома Ангельмана.
Формы
Синдром Ангельмана связан с четырьмя вариантами генетических мутаций:
- Вновь возникшей хромосомной мутацией, которая связана с потерей участка хромосомы в локусе 15 q11 — q13 . Данная мутация – причина около 80 % всех случаев заболевания.
- Одноотцовской дисомией, которая связана с потерей материнского локуса (отсутствие генетического материала матери). Данный вариант встречается редко (около 5% всех случаев).
- Дефектом ряда генов, подверженных геномному импринтингу (ГИ). Данные дефекты возникают у 2-4% больных в результате непосредственного нарушения импринтинга (различия в преобразовании информации гена в белок или РНК, которые зависят от происхождения гена). Чаще всего возникает в результате утраты центра регуляции ГИ. Дефекты ГИ без утраты центра регуляции являются результатом спонтанной мутации, повторение которой – большая редкость.
- Спонтанной мутацией материнской копии, которая вызывает отсутствие преобразования в мозге копии гена UBE3A. Данный ген кодирует деятельность убиквитинлигазы (фермент, участвующий в сложном процессе распада белков). Дефицит данного фермента относится к молекулярным механизмам синдрома.
Установить форму заболевания у 7-9 % в настоящее время не представляется возможным.
Синдром Ангельмана – что это такое?
Данная патология является врожденным заболеванием. Она развивается внутриутробно. Это нарушение впервые описал педиатр из Великобритании – доктор Гарри Ангельман в 1965 году. Тогда пациентов с таким диагнозом называли «кукольными детьми». Сама же болезнь именовалась «синдромом счастливой марионетки». У данного заболевания есть и другие названия. Одно из них – синдром Петрушки. Данная патология сопровождается задержкой физического и умственного развития, хождением на прямых ногах и смехом без всяких на то причин. Внешне пациенты с таким диагнозом похожи на кукол.
Синдром Ангельмана – причины
По сей день ученые изучают факторы, провоцирующие развитие этой патологии. Чаще развивается синдром Ангельмана, генетика если нарушена. Другими словами, вероятность его возникновения возрастает, когда кто-то из родителей имеет хромосомные дефекты. Однако синдром Ангельмана, синдром Петрушки, смеющейся куклы появиться может и спонтанно у малыша, родители которого совершенно здоровы.
Тератогенное воздействие на ребенка оказывают следующие факторы:
- вредные привычки беременной;
- длительный прием родителями психотропных препаратов;
- сильные переживания будущей мамы, спровоцированные горем или чрезмерным стрессом;
- воспалительный процесс, поразивший репродуктивную систему беременной;
- пребывание будущей мамы в зоне повышенного радиационного излучения.
Синдром Ангельмана – тип наследования
Данная патология возникает из-за нарушения в процессе деления хромосом. Синдром Ангельмана кариотип имеет 46 XX или XY. Дефекты обнаруживаются в 15-ой хромосоме. Мутация проявляется следующим образом:
- Делеция
– выпадение частички хромосомы. Пациенты, имеющие такое нарушение, страдают сильной умственной отсталостью. Эти дети в большинстве случаев не умеют ходить и говорить. К тому же таких пациентов часто одолевают эпилептические припадки. - Дупликация
– появление лишних генов. Если диагностирован такой синдром Ангельмана, пациенты чаще умирают в младенческом возрасте. Иногда такие люди доживают до полового созревания. - Инверсия
– дефект, при котором отсутствует часть хромосомы. При этом гены расположены в обратной последовательности. - Транслокация
– часть хромосомы присоединена к другой. - Однородительская дисомия
– нарушение, при котором ребенок наследует две копии отцовского набора 15-ой хромосомы. Материнскую же он не получает.
Этиология заболевания
Данный синдром встречается довольно редко, примерно в 1 случае на 10-20 тысяч новорожденных. Основной причиной заболевания является потеря копии нормальных генов матери в 15 хромосоме, что происходит вследствие нарушения процесса деления этой хромосомы. Помимо этого, заболевание может возникнуть вследствие мутации генов по линии отца, отцовской трисомии или дисомии.
В норме у здорового человека наблюдается передача по одной копии 15 хромосома от матери и от отца. Если же ребенок получит от одного из родителей несколько генетически измененную копию (особенно если это ген матери, поскольку они более сильные по сравнению с генами отца), то у малыша будет развиваться синдром Ангельмана.
Классификация и симптомы патологии
Клиническая картина поражения во многом определяется видом генетического дефекта, который обуславливает его возникновение. В медицине принято различать несколько типов болезни:
- Потеря участка 15-ой хромосомы – самая распространенная форма кариотипа, приводящая к возникновению симптомов при синдроме Ангельмана. Она диагностируется в 80% случаев.
- У 5% пациентов отмечается утрата части материнской генетической информации. Участок 15-й хромосомы, которая должна быть получена от женской яйцеклетки, заменяется материалом из ДНК сперматозоида. Данное явление известно, как отцовская дисомия.
- Мутировать могут сразу несколько генов. Такая проблема регистрируется лишь в 3–4% случаев. Синдром Ангельмана у детей с подобным дефектом обусловлен нарушениями процесса импринтинга. Это происходит в результате мутации генов, контролирующих данный механизм.
- У некоторых пациентов отмечается спонтанное формирование дефекта. В таких случаях осуществляется преобразование гена, расположенного в головном мозге ребенка. Эта структура отвечает за ферментацию белков. Симптомы формируются на фоне дефицита данного соединения.
В 6–8% случаев определить тип поражения не удается.
Синдром Ангельмана сопряжен с различными клиническими проявлениями, включающими в себя:
- Отставание в умственном и физическом развитии. Специфическим симптомом является уменьшение объема черепа. Ребенок с синдромом Петрушки плохо поддается обучению, поскольку болезнь связана с нарушением работы головного мозга.
- Характерным признаком поражения является то, что малыш склонен постоянно улыбаться и смеяться. При этом поводы для подобного поведения зачастую отсутствуют. Пациенты выглядят жизнерадостными и охотно идут на контакт с другими детьми и взрослыми. Именно подобная манера позволяет врачам заподозрить наличие нарушения.
- Нередки и неврологические симптомы. Они включают в себя тремор, гиперактивность верхнего пояса конечностей, а также специфическую походку. Дети передвигаются таким образом, что ноги остаются прямыми. Это и дало первоначальное название патологическому состоянию – синдром счастливой куклы, или марионетки. У некоторых пациентов регистрируются судороги, ухудшающие дальнейший прогноз.
- Речевая дисфункция – распространенная проблема у детей с синдромом Ангельмана. При этом врачи зачастую отмечают хорошее понимание слов окружающих, сочетающееся с затрудненным выражением собственных мыслей. Проблема накладывает серьезный отпечаток на социальную жизнь пациентов.
- Возможны и деформации черепа, его мозгового и лицевого отдела. У детей выдвинутый вперед массивный подбородок, широкий рот, а также аномалии закладки и развития зубов.
Характерно то, что усиление симптомов в среднем приходится на возраст 5–7 лет. У новорожденных клиническая картина в большинстве случаев неспецифична и не позволяет заподозрить синдром. Специфические нарушения, подтверждающие диагноз, отмечаются в дошкольный период и зачастую ассоциированы с отставанием от сверстников в умственном и физическом развитии.
Основной проблемой при заболевании остается снижение коммуникативных навыков, а также трудности при самообслуживании. Около 80% пациентов с синдромом Ангельмана выражает свои мысли посредством 3–4 слов. Дети и взрослые с генетической аномалией хорошо объясняются при помощи жестов, поэтому широко используют их в повседневной жизни. Данная характеристика лишь доказывает относительно высокие интеллектуальные способности. Формальные языки остаются труднодоступными, вследствие чего методики взаимодействия с окружающими людьми индивидуальны в каждом случае. Доказано, что пациенты с синдромом Энгельмана лишены возможности мыслить абстрактно.
Навыки самообслуживания также варьируются. Подавляющее большинство людей с генетической аномалией способны самостоятельно ходить, справлять нужду, пользоваться столовыми приборами. Пациенты учатся сообщать о предпочтениях, касающихся еды. Им может потребоваться помощь при купании, а также использовании одежды с пуговицами и молниями. При этом многие действия, совершаемые больными в повседневной жизни, требуют контроля. Такая необходимость обусловлена тем фактом, что у людей с синдромом Ангельмана полностью отсутствует чувство опасности. Однако при должной поддержке со стороны близких они хорошо адаптируются к внешним условиям.
Аномалии генетического материала
Наследственный материал состоит из огромного количества нуклеотидов, формирующих гены. При этом в каждом гене последовательность нуклеотидов строго определена, поскольку должна кодировать определенный белок. Кроме того, сами гены при формировании хромосом также выстраиваются в фиксированном порядке. Благодаря сохранению этого порядка организм может функционировать, а ученые – быстро и точно указывать друг другу, про какой ген идет речь.
В идеальном случае система работает без малейших сбоев, а генетическая информация всегда передается в неизменном виде. Однако на практике большое число структурных единиц и постоянное воздействие различных факторов (например, ионизирующего излучения) приводит к тому, что время от времени возникают различные аномалии. В частности, отдельные участки последовательности ДНК могут быть скопированы на новое место. В таком случае говорят о дупликации. Если же вместо создания новой копии была перемещена часть исходной цепочки, то модификация называется транслокацией. Кроме того, иногда часть последовательности просто теряется, удаляется из генетического материала. В таком случае изменение называется делецией.
Поскольку взаимодействия в организме оттачивались в течение многих тысячелетий эволюционного развития, получилась очень слаженная система. И аномалии, даже самые небольшие, могут вызвать нарушение баланса. В таком случае в организме развивается то или иное нарушение. Если при этом причина находится на уровне генов, то говорят о генных болезнях. Если была утрачена или наоборот получена лишняя копия хромосомы, то такие нарушения называются хромосомными заболеваниями.
Примечания
- X-сцепленная умственная отсталость. (рус.). Центр Молекулярной генетики при Медико-генетическом научном центре РАМН. Проверено 8 мая 2021.
- Yamasaki K., Joh K., Ohta T., Masuzaki H., Ishimaru T., Mukai T., Niikawa N., Ogawa M., Wagstaff J., Kishino T.
Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. (англ.) // Human molecular genetics. — 2003. — Vol. 12, no. 8. — P. 837—847. — DOI:10.1093/hmg/ddg106. — PMID 12668607. - Petersen M. B., Brøndum-Nielsen K., Hansen L. K., Wulff K.
Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: estimated prevalence rate in a Danish county. (англ.) // American journal of medical genetics. — 1995. — Vol. 60, no. 3. — P. 261—262. — DOI:10.1002/ajmg.1320600317. — PMID 7573182. - Steffenburg S., Gillberg C. L., Steffenburg U., Kyllerman M.
Autism in Angelman syndrome: a population-based study. (англ.) // Pediatric neurology. — 1996. — Vol. 14, no. 2. — P. 131—136. — DOI:10.1016/0887-8994(96)00011-2. — PMID 8703225.
Лечение
Синдром Ангельмана обусловлен врожденной генетической аномалией; в настоящее время специфические способы его лечения не разработаны, однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом. Младенцы с гипотонусом должны получать массаж и другие виды специальной терапии (физиотерапии).
Рекомендуются использование специальных методик развития ребенка, занятия с логопедом и дефектологом. Нарушения сна корректируются назначением легких снотворных. Д-р Вагстафф (США) считает, что назначение 0,3 мг мелатонина за 0,5–1,0 ч перед сном улучшает сон пациентов с синдромом Ангельмана. Нарушения стула регулируются назначением легких слабительных.
При судорожных припадках подход к лечению такой же, как при эпилепсии. У детей с синдромом Ангельмана часто наблюдаются приступы более чем одного типа. Показана электроэнцефалография.
Требует коррекции нежелательное поведение. Д-р Чарльз Вильямс (Гейнсвилл, Флорида), работающий в основном с аутичными детьми, отмечает общие для аутичных детей и детей с синдромом Ангельмана особенности поведения: заметная аутостимуляция, импульсивность, навязчивые, повторяющиеся движения, интерес к неуместным предметам, а также сложности в общении с другими людьми. Врачи США сообщают, что у аутичных детей внутривенные инъекции гормона секретин (найденного в поджелудочной железе) успешно уменьшают проявления нежелательного поведения и обеспечивают хороший уровень общительности и коммуникативных навыков; возможно, медицина придет к использованию секретина для коррекции поведения детей с синдромом Ангельмана.
Джованни Франческо Карото (ок. –). Портрет рыжеволосого мальчика с рисунком шарнирной куклы
Прогноз
Прогноз при синдроме Ангельмана зависит от характера хромосомной аномалии и своевременности ее обнаружения. Тяжелее всего приходится тем деткам, чья 15 хромосома содержит «пропуски» генов (делеция). Вероятность ходить и разговаривать у таких пациентов чрезвычайно мала. Остальные случаи при внимательном подходе и любви к своему ребенку поддаются коррекции.
Стать полноценными членами общества такие больные, увы, не смогут, при всем том, что они далеко не глупы, понимают речь и ее смысл. Вот только проблемы с общением у них остаются на всю жизнь. Пациентов с детства можно обучить языку жестов, но нельзя заставить общаться при помощи слов. Лексикон «говорящих» больных ограничивается минимумом слов, употребляемых в быту (5-15 слов).
Что касается продолжительности жизни и общего состояния здоровья больных с синдромом Ангельмана, то здесь цифры колеблются на средних показателях. Во взрослой жизни пациенты в основном сталкиваются с такими проблемами со здоровьем, как сколиоз и ожирение, которые при правильном подходе к лечению не опасны для жизни.
(
2 оценок, среднее: 5,00 из 5)
Синдром Ангельмана причины
При данном заболевании отсутствуют отдельные гены 15-й хромосомы. Большинство случаев характеризуется частичной делецией или же мутацией 15-й хромосомы. При синдроме Ангельмана страдает материнская хромосома, а в случае изменений в отцовской хромосоме развивается синдром Прадера — Вилли. Зачастую синдром провоцируется спонтанным хромосомным дефектом, который характеризуется отсутствием смежной области, состоящей из четырех миллионов пар оснований ДНК в местах q11- q13 15-й хромосомы.
Синдром Ангельмана имеет кариотип 46 XX или XY, 15р. Независимые исследования к причинам синдрома Ангельмана относят мутации в гене UBE3A. Ферментный компонент сложной системы деградации белков считают продуктом этого гена.
Диагностика
Чаще всего диагноз ставят, когда ребенок достигает возраста 3-7 лет. В этот период появляются симптомы недуга, по которым предварительно и определяют его наличие.
В период новорожденности выявить патологию достаточно сложно, так как клинические проявления отсутствуют, имеют скрытый характер. Помимо анализа признаков заболевания.
Проводят ряд исследований, таких как определение тонуса мышечной ткани, а также генетического исследования 15 хромосомы. Нарушения данного элемента свидетельствуют о наличии синдрома Ангельмана.
Как лечат синдром Ангельмана
Это генетическое заболевание и, к сожалению, ученые еще не нашли методов его лечения. Но существуют методики, которые облегчают жизнь таким людям и значительно повышают качество их жизни.
В каждом индивидуальном случае при таком заболевании, как синдром Ангельмана у детей, симптомы будут иметь свои отличия. Поэтому для каждого пациента разрабатывается индивидуальная методика терапии.
Существует несколько типов терапевтического лечения, они заключаются в следующем.
- Так как для синдрома Ангельмана характерны припадки, конвульсии, приступы эпилепсии, рекомендованы противоэпилептические препараты, или антиконвульсанты. Они значительно снизят проявления этих симптомов и позволят контролировать состояние пациента.
- Нарушение моторики, проблемы с двигательными функциями также являются симптомами синдрома Ангельмана. Занятия лечебной физкультурой и специальные тренировки, разработанные для таких пациентов, необходимы для их развития. Нужно регулярно и терпеливо выполнять упражнения, и тогда работа будет вознаграждена. Такие дети медленно развиваются, но благодаря терпению родителей можно многого добиться.
Специальные занятия помогают деткам с синдромом Ангельмана адаптироваться к внешнему миру
Деткам с гипотонусом должен быть назначен массаж и специальные физиотерапевтические процедуры. Если у ребенка синдром Ангельмана, лечение обязательно должно включать язык жестов. Это будет очень полезно, как для них в дальнейшем общении с людьми, так и для самих мам и пап. Рекомендованы занятия с дефектологом и логопедом. Специально разработанные программы помогают корректно и эффективно учить детей правилам этикета и поведению в обществе
Они рассчитаны на то, чтобы помочь пациентам справиться с повышенной активностью, научиться концентрировать свое внимание. После прохождения такой терапии пациентам будет легче общаться со сверстниками, и некоторые даже могут создавать свои семьи. Надо тщательнее подходить к составлению рациона питания, так как у таких детей очень часто наблюдается ожирение, особенно у девочек.
При наличии синдрома Ангельмана нарушаются обменные процессы в организме, что часто может приводить к избыточной массе тела при нормальном питании
- У таких пациентов, как правило, нарушен стул, в этом случае назначают слабительные.
- При нарушении сна назначают легкие снотворные.
Ученые заметили, что синдром Ангельмана очень схож с таким заболеванием, как аутизм.
Клиническая картина в подробностях
У разных больных могут проявляться разные симптомы. Эта разница зависит от степени и вида хромосомного отклонения.
Можно разделить признаки по частоте их проявления у детей, больных синдромом Ангельмана:
- Симптомы, которые проявляются у всех больных. К ним относят тяжелую функциональную задержку развития, отклонения в поведении (смех и улыбка, не имеющие причины, состояние счастья, повышенная возбудимость, пониженная концентрация внимания). Также у абсолютно всех пациентов наблюдается нарушение моторных функций, нарушение равновесия, тремор, преобладание невербальных навыков над словесными, нарушения речи.
- Симптомы, характерные для 80% пациентов. Задержка в росте, приводящая к диспропорции головы относительно других частей тела. Зачастую это приводит к развитию микроцефалии. У детей до трех лет часто проявляются эпилептические приступы, затем они становятся реже либо исчезают вовсе. Результаты электроэнцефалограммы часто аномальны, волны низкого уровня имеют повышенную амплитуду и временную динамику.
- Симптомы, возникающие у менее чем 80% больных. К таким проявлениям относят косоглазие, пониженную способность контролировать движения языка, проблемы с глотанием, гипопигментацию глаз и кожи, альбинизм, У многих пациентов наблюдается повышенная активность сухожильных рефлексов. В раннем детстве у многих возникают проблемы с питанием и сном. Среди проявлений также можно назвать слюнотечение, высовывание языка, постоянная жажда. К внешним признакам относят также наличие плоского затылка и гладких ладоней.
По мере взросления проявления синдрома изменяются. Реже наблюдаются расстройства сна, гиперактивность, судорожный синдром. Взрослые, имеющие синдром Ангельмана, выглядят моложе своих ровесников.
Половое созревание происходит немного позже, чем у индивидов, не имеющих данной патологии. Больные могут иметь собственных детей, однако высок риск передать заболевание потомству.
Многие взрослые пациенты страдают от неконтролируемого мочеиспускания. Часто встречается наличие сложностей с моторикой, что приводит к необходимости носить одежду без молний, пуговиц. Среди взрослых больных наблюдается проблема лишнего веса, поэтому необходимо контролировать соблюдение специальной диеты.
Дети хорошо воспринимают устную речь, и понимают содержание почти всех разговоров, однако редко отвечают. Часто они отказываются принимать участие в разговоре, и употребляют в речи несколько десятков слов.
Симптомы синдрома Ангельмана
Проявления расстройства в психо-эмоциональной сфере:
- задержка развития, в том числе отсутствие лепета и ползания в период от 6 месяцев до 1 года;
- угнетенная обучаемость;
- отсутствие речи либо минимальный навык говорения;
- немотивированный частый смех/улыбка;
- тип личности можно описать как радостный, экзальтированный.
Симптомы расстройства на соматическом уровне:
- приступы эпилепсии, которые обычно возникают в возрасте от 2 до 3 лет;
- затрудненная ходьба/перемещение, скованные или резкие угловатые движения из-за неспособности контролировать произвольные движения (атаксия);
- необычное поведение, такое как внезапные взмахи рук, ходьба с приподнятыми руками и/или на негнущихся ногах;
- небольшой размер головы с уплощением в затылочной зоне (микробрахицефалия);
- волосы, кожа и глаза светлого оттенка (гипопигментация);
- иногда у детей наблюдаются широкий рот, редкие зубы и высунутый язык. В большинстве случаев черты лица пациентов не отличаются чем-то необычным.
Отдельно следует остановиться на особенностях речевого развития и моторики, характерных для людей с синдромом Ангельмана.
У большинства детей с этим расстройством отмечают крайне скудный словарный запас, который может быть ограничен несколькими словами (до одного-двух десятков). Это обусловливает значительные трудности социализации ребенка, у которого наблюдается синдром Петрушки.
При этом дети, имеющие такое расстройство – как правило – понимают не только простые команды, но и способны усваивать информацию в объеме, который значительно превышает их возможности общения и выражения мыслей.
Что касается моторики, то кроме описанных выше проявлений, дети-«Петрушки» могут часто держать руки приподнятыми и согнутыми в локтях и запястьях, и периодически взмахивать руками в моменты возбуждения или во время ходьбы. Именно эта схожесть с движениями марионетки в купе с их угловатостью и скованностью на фоне частой экзальтированности пациентов стала причиной для названия расстройства синдромом счастливой куклы.
У них может наблюдаться как пониженный мышечный тонус туловища (гипотония), так и увеличенный мышечный тонус конечностей (гипертония), а также аномально преувеличенные рефлекторные реакции (гиперрефлексия). У некоторых детей развивается мелкий тремор рук и ног. Эти нарушения могут проявиться в возрасте от 6 месяцев до 1 года.
Освоение некоторых этапных навыков развития моторики – например, ходьбы – происходит с задержкой.
В случаях относительно легкой формы болезни, дети могут начать ходить в 2-3 года. В более тяжелых случаях прямохождение замедлено и происходит рывками – «ригидно», что тоже напоминает движения марионетки. Некоторые дети не ходят до возраста от 5 до 10 лет. Примерно 10% пациентов не способны перемещаться без посторонней помощи.
Влияние делеций на способность к оплодотворению
Делеции, происходящие в обычных хромосомах (аутосомах) могут в некоторых случаях быть компенсированы нормальной копией гена. Однако когда речь заходит о половых хромосомах, особенно об Y-хромосоме, ситуация меняется.
Прежде всего необходимо отметить, что локализованные на ней гены не имеют второго экземпляра. При нормальном количестве хромосом в наборе Y-хромосома оказывается крайне уязвимой. В сочетании с малым количеством генов это приводит к серьезным последствиям каждого изменения. Особый интерес представляют мутации, касающиеся AZF-локуса и SRY гена.
Лечение
Профилактики и избавления от синдрома петрушки (куклы) современными средствами не существует. Лечения синдрома Ангельмана сегодня не существует из-за того, что неизвестны причины генетического дефекта. Разработаны комплексы мероприятий, направленные на стабилизацию и улучшение качества жизни при таком заболевании. Программа реабилитации разрабатывается индивидуально, но некоторые из мер являются общими для всех больных:
- Прием антиконвульсантов и противоэпилептических средств.
- Лечебная профилактическая физкультура, массаж, физиотерапия. Помогает стабилизировать навыки общей моторики, наладить работу опорно-двигательного аппарата, учить ходить естественно(на фото).
- Изучение языка жестов. Многие больные синдромом петрушки используют в разговоре набор слов меньше среднего, но при этом нет сложностей с обучением. Язык жестов становится приемлемой альтернативой в общении, если обучать ему с раннего детства(на фото).
- Терапия поведения. Данный метод направлен на компенсацию дефицита внимания, устранение или подавление гиперактивности, возможность дать корректное воспитание для адаптации в социуме.
Профилактика
Предупредить рождение ребенка с синдромом Ангельмана невозможно, поскольку мутация генов происходит спонтанно. Объяснить подобный процесс не под силу даже самым квалифицированным генетикам.
Профилактические мероприятия направлены на тщательную подготовку к беременности и рождению ребенка. Все беременные женщины должны пройти в назначенный срок УЗИ плода и сдать кровь на сывороточные маркеры хромосомных патологий. В случае положительного результата медико-генетического исследования родители должны решить, смогут ли они воспитывать больного ребенка.
Медико-генетическое консультирование требуется парам с отягощенной наследственностью. Если данное заболевание не было зафиксировано у близких и родных, достаточно соблюдать основные принципы ведения здорового образа жизни во время беременности.
Синдром Ангельмана – тяжелая и неизлечимая болезнь, лишающая человека нормальной жизни. Больные дети нуждаются в особой заботе и внимании. Лечение направлено на снижении интенсивности симптомов патологии. Современные ученые стараются найти действенные способы борьбы с хромосомными аномалиями, жертвами которых становятся новорожденные дети.