Craniopharyngioma is a rare type of brain tumor. It is localized in the pituitary gland. It is mainly first diagnosed in children aged 5-14 years or in adults over 40. The survival rate for craniopharyngioma for 10 years is about 90%. For a long time, the neoplasm does not manifest itself with any symptoms, but over time, multiple abnormalities develop: endocrinological, visual, hormonal.
Reasons for development
Craniopharyngioma develops due to excessive proliferation of epithelial cells in Rathke's pouch. It is formed in the fetus in the first trimester of pregnancy and is the basis for the future pituitary gland. If development occurs normally, then after the formation of the pituitary gland, Rathke cells are no longer produced. Craniopharyngioma appears only if this tissue continues to divide.
Typically, pituitary craniopharyngioma occurs due to a hereditary predisposition. A mutation of chromosomes occurs, which leads to the formation of a tumor. It can also occur due to intrauterine development disorders in the first trimester of pregnancy - this is when all the fetal organs are formed. This can be affected by taking medications, exposure to radiation, poisons and toxins.
Intrauterine infection and early severe toxicosis significantly increases the likelihood of craniopharyngioma. Chronic diseases of the mother, such as kidney pathologies, diabetes mellitus or tuberculosis, can contribute to the development of a tumor in a child.
Craniopharyngioma
18.11.2015
Craniopharyngioma is a benign tumor that develops from the remains of Rathke's pouch epithelial cells. Rathke's pouch is formed in the 4th week of gestation in the form of a protrusion of the pharyngeal membrane and in the 5th week it connects with a growth of the neural tube, which subsequently forms the neurohypophysis. The adenohypophysis forms from the ventral part of Rathke's pouch, and the proximal part (craniopharyngeal duct) is normally obliterated at the 7th week of gestation. In 25–70% of cases, according to the literature, autopsies reveal remnants of Rathke's pouch cells, which can be a source of growth of craniopharyngiomas.
Craniopharyngioma accounts for about 9% of all tumors of the central nervous system and about 56% of all tumors of the chiasmal-sellar region. It is the third most common brain tumor in children after medulloblastoma and astrocytoma.
In the clinical picture of the disease in children, three leading syndromes are distinguished:
- general cerebral (headache, vomiting, impaired consciousness);
- visual disturbances (decreased visual acuity, narrowing of visual fields);
- endocrine disorders (hypopituitarism, diabetes insipidus, diencephalic disorders).
Currently, the treatment of choice for patients with craniopharyngioma is radical tumor removal. Most operations are performed transcranially, in some cases transnasal removal is possible. If it is impossible to radically remove a craniopharyngioma (with a complex anatomical and topographical variant and in the treatment of relapses), it is advisable to supplement partial tumor removal with external beam radiation therapy or perform puncture treatment. After tumor removal in children, a significant proportion of cases develop a deficiency of pituitary tropic hormones.
Clinical case:
Patient M. has been observed at the Federal State Budgetary Institution Research Center since the age of 7 years.
The boy was born from a normal pregnancy, at term, with normal weight and height (50 cm, 3450 g, Apgar score at birth 8–9 points). Early development proceeded without any special features. There is no hereditary history of endocrinopathies; the patient’s parents and close relatives did not have any neoplasms.
At the age of 5 years, against the background of complete health, the child first began to complain of headaches. During the examination by a neurologist, an MRI of the brain was not performed at the site; according to the clinical examination, no evidence of CNS pathology was identified; taking NSAIDs was recommended in case of headaches. Over the next 2 years, the child periodically complained of headaches, but after taking NSAIDs and sleeping, the pain went away.
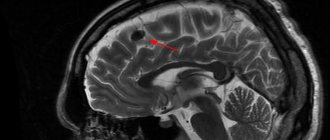
At the age of 7 years, the headaches increased significantly. A month later, the child, while at home, suddenly lost consciousness, then tonic-clonic convulsions developed, which were stopped by the ambulance after the administration of diazepam. The boy was urgently hospitalized in the neurological department, where an MRI of the brain revealed a cystic craniopharyngioma of the chiasmatic-sellar region, occlusive hydrocephalus with periventricular edema. When examined by an ophthalmologist, a significant decrease in visual acuity was revealed, which indicated compression of the optic chiasm by a space-occupying formation.
The child was urgently consulted at the Research Institute of Neurosurgery named after. N.N. Burdenko, emergency surgical treatment was recommended.
Considering the fact that most of the tumor was represented by a cystic component, the patient had a catheter installed in the suprasellar cyst of the craniopharyngioma and connected to the subcutaneous Ommaya reservoir, followed by aspiration of the cystic contents.
The postoperative period was satisfactory. The patient did not have postoperative electrolyte disturbances in the form of hypernatremia, and there were no pronounced manifestations of diencephalic syndrome (impaired thermoregulation, sleep disturbances, eating behavior, memory impairment, mental disorders).
However, given the location of the tumor and its close proximity to the pituitary gland, to exclude hypopituitarism, the patient was recommended to be examined at the Federal State Budgetary Institution Endocrinological Center.
The boy was first examined at the Research Institute of Pediatric Endocrinology of the Federal State Budgetary Institution Endocrinology Center at the age of 7 years. On examination, height is 118.3 cm (height SDS -1.36), weight is 25 kg, growth rate is within the age norm. The weight gain in the postoperative period was noteworthy (2 kg in 2 months).
Based on the results of the examination, secondary hypothyroidism (st. T4 16.36 pmol/l) and secondary hypocortisolism (serum cortisol 643.9 nmol/l) were excluded. The boy did not complain of polyuria and polydipsia, which, together with high specific gravity of urine (1014–1024 g/l), normal daily diuresis (1040–1100 ml/day) according to urine analysis according to Zimnitsky, as well as the absence of electrolyte disorders (blood sodium level 135–139 mmol/l) made it possible to exclude the presence of diabetes insipidus.
One of the most common complications of surgical interventions for formations of the chiasmatic-sellar region is the development of acquired GH deficiency. This complication most often manifests itself as a decrease in growth rates in the postoperative period. However, taking into account such an early examination of the patient after surgery, the growth rate did not have time to decrease and the boy’s height was within the age norms (height SDS -1.36). IGF-1 level is 64.13 ng/ml. Bone age was one year behind the passport age and corresponded to 6 years. To assess the preservation of GH secretion by the pituitary gland, a GH stimulation test with clonidine was performed. The maximum release of growth hormone in the test was 3.8 ng/ml, which confirmed the presence of GH deficiency (stimulated release of growth hormone less than 10 ng/ml). Daily injections of recombinant growth hormone were recommended at a rate of 0.033 mg/kg/day.
The issue of prescribing rGH drugs in the early postoperative period remains controversial. However, convincing evidence has now been obtained that there is no continued growth of residual craniopharyngioma tissue during therapy with rGH drugs. That is why, taking into account the patient’s age, poor growth prognosis and the high probability that the patient will not achieve a socially acceptable final height, after consultation with a neurosurgeon, it was decided to prescribe rGH drugs. It is worth remembering that after prescribing rGH drugs in the postoperative period, annual follow-up is necessary with mandatory MRI of the brain and examination by an ophthalmologist (assessment of the condition of the fundus, assessment of visual fields).
During therapy with growth hormone drugs, the patient's growth rate remained satisfactory; the growth rate averaged 10 cm per year, which corresponded to the age norm. By following dietary recommendations and using feasible physical activity, it was possible to normalize the patient’s weight. With regular monitoring of MRI of the brain (once a year), no evidence of growth of residual tumor tissue was obtained. There is no need for additional emptying of a craniopharyngioma cyst. Normalization of visual acuity is noted (Vis OS = OD = 1.0).
Conclusion:
Currently, the use of highly informative imaging techniques makes it possible to diagnose brain space-occupying lesions in the early stages. If a child complains of headaches, decreased visual acuity, the appearance of convulsions and loss of consciousness, a mandatory consultation with a neurologist is necessary, an MRI of the brain is required, which is the most accurate method for diagnosing craniopharyngioma, identifying its location and relationship with brain structures. All patients with space-occupying formations in the chiasmatic-sellar region undergo not only an ophthalmological examination with assessment of the fundus, but also mandatory perimetry with assessment of visual fields. Advances in modern neurosurgery and neuroreanimatology make it possible to radically remove craniopharyngiomas in more than 80% of patients without causing severe neurological complications. In the presence of a cystic form of the tumor with a minimal dense part, it is recommended to install an Ommaya type system with subsequent aspiration of the cystic contents.
An important aspect of patient management after surgical removal of space-occupying formations of the chiasmatic-sellar region is the timely diagnosis of postoperative hypopituitarism and adequate replacement therapy, which can significantly improve the quality of life of operated patients.
Craniopharyngioma in children
Craniopharyngioma is uncommon in children. It is diagnosed in only 4% of all brain tumors. Typically, the first signs of the disease are diagnosed at the age of 5-14 years; craniopharyngioma occurs equally often in boys and girls. Initially, the pathology does not manifest itself with any obvious signs. Over time, the patient begins to complain of the following symptoms:
- Frequent headaches, worse in the morning;
- Nausea and vomiting;
- Delayed sexual and physical development;
- Violation of hormonal and endocrine functions;
- Change in behavior;
- Confusion;
- Visual impairment;
- Decreased appetite, increased thirst;
- Impaired motor functions;
- Increasing epilepsy;
- Swelling of the fundus;
- Altered psychopathological state.
Depending on the location of the tumor and its size, the manifestations of the disease may vary. It is important for parents to monitor the condition of their child, because timely diagnosed craniopharyngioma responds well to treatment.
Symptoms of craniopharyngioma
The clinical picture of craniopharyngioma may not manifest itself for a long time or may appear in the form of weak signs characterizing the growth of the tumor. The symptoms of this type of tumor are often of a purely individual nature and depend on many factors, primarily on the characteristics of the patient’s body and his chronic diseases. Vivid symptoms most often appear in patients aged ten to twenty years. More than half of these tumors are cystic neoplasms, just over ten percent are solid tumors, and there are also mixed-type craniopharyngiomas.
If a person develops craniopharyngioma of the brain, then, first of all, disturbances occur in the endocrine system (disturbances in the functioning of the adrenal glands, diabetes insipidus, menstrual cycle disorders in women, potency in men). Pain syndromes are also observed, vision decreases, signs of hydrocephalus may occur (due to impaired transport of cerebrospinal fluid in the third ventricle), as well as neuropsychological disorders. In adolescence, with the development of a tumor of this type, puberty is delayed or, conversely, its development is too early and very rapid.
Headache, which is one of the first signs of brain tumors, in this case is characterized by an increasing character. Visual impairment is associated with the growth of the tumor body, which compresses the optic nerve. Over time, entrapment of the optic nerve can lead to complete atrophy.
Neuropsychological disorders may manifest as dementia, bulimia, or severe to moderate obesity. Retardation, deterioration of motor skills, emotional instability, apathy, and impaired short-term memory may also be observed.
In children, symptoms of craniopharyngioma are most often expressed in growth retardation.
Craniopharyngioma in adults
Craniopharyngioma is a benign tumor in the brain. Usually it begins during embryonic development and is localized in the hypothalamic-pituitary region. Due to the growth of the tumor, numerous cysts often form, inside which fluid from proteins and cholesterol accumulates. In this case, the papillary form of craniopharyngioma is mainly diagnosed in adults.
Suprasellar and intrasuprasellar craniopharyngioma
The initial classification of craniopharyngiomas was developed by V. Grekhov at the Institute of Neurosurgery named after. Burdenko. In the future, it was repeatedly detailed and changed. Today it is customary to distinguish the following topographic variants of craniopharyngiomas:
- Suprasellar
. Such tumors grow within the sella turcica, displacing the diaphragm. They can spread supra- and parasellar. The neoplasm develops from the remnants of the epithelium, which is preserved at the level of the pituitary gland; - Stem
. They spread in the area of the pituitary gland stalk, gradually affecting the bottom of the 3rd ventricle. They are localized above the sella turcica, displacing its diaphragm down; - Intra-extraventicular
. They develop at the level of the pituitary infundibulum and spread into the cavity of the third ventricle. This type of craniopharyngioma is associated with the accumulation of embryonic epithelium in the infundibulum area.
Adamantimatous and papillary craniopharyngioma
Craniopharyngiomas vary in structure. They may contain cysts or consist of dense tissue. Cystic formations are characterized by slow growth, extremely rarely their diameter exceeds 5 centimeters. Typically, such tumors have up to 50 cysts, in rare cases their number reaches 200. The formation itself is dense, with a strong capsule, and quickly connects with brain tissue and blood vessels.
NB!
Under the influence of various factors, the tissue structure of craniopharyngioma can change.
If craniopharyngioma has been present in the body for a long time, there is a risk of necrosis. This contributes to the rapid formation of cysts. Inside they are filled with liquid with a high lipid content. Craniopharyngiomas can vary in cytological structure. The following types of such tumors are distinguished:
- Adamantine-like
. Occurs in most cases. Mostly diagnosed in children. A neoplasm of this type consists of remnants of embryonic tissue. They mainly consist of cysts. Adamantine-like craniopharyngiomas are characterized by polymorphism; - Papillary
. It is mainly diagnosed in people over 40 years of age, and practically never occurs in children. Such a tumor consists of metaplastic cells, with virtually no fossilization. Papillary craniopharyngiomas can consist not only of epithelial but also epidermal cells. In such tumors, degenerative processes occur and cysts and stroma develop.
Epidemiology and embryology
Among the total number of primary brain tumors, craniopharyngiomas account for 1.2-4.6%. In patients under 16 years of age, the incidence of tumors ranges from 5-10%, and in adults it can reach up to 60%. There are two age peaks for the high probability of developing craniopharyngiomas: in children from 6 to 16 years and in adults from 48 to 75 years. Every year, from 0.5 to 2 cases of this type of tumor are diagnosed per 1 million population.
The note. Most neoplasms of this type are diagnosed in China (up to 6.5% of cases), least of all in Australia (up to 1.5%).
According to the theory proposed by Erdheim (it has greater scientific weight), craniopharyngiomas in the brain are formed from the remnants of the tissue structure of the epithelial embryonic tissue of Rathke's pouch, which is an invagination of the primary oral tube. From this structure, at the initial stages of embryogenesis, the anterior lobe of the pituitary gland, as well as its tuberal region, is formed.
Symptoms
Craniopharyngioma of the pituitary gland is characterized by the development of endocrine and metabolic disorders, and intracranial hypertension syndrome. Because of this tumor, the balance of many hormones in the body is disrupted: gonadotropin, LH, ACTH, STH. This leads to the development of hypothyroidism, hypogonadism, hypocortisolism, and diabetes insipidus. Over time, hypothalamic-pituitary disorders occur, such as:
- Constant feeling of thirst;
- Polyuria, enuresis;
- Developmental delays, decreased growth rates in children;
- Rapid increase in body weight;
- Increased fatigue, muscle weakness;
- Menstruation irregularities, decreased libido.
The severity of such deviations depends on the location of the craniopharyngioma, its size and the age of the patient. Early symptoms of a tumor also include visual disturbances: decreased visual acuity, development of blindness. If the tumor is located in the third ventricle and compresses its openings, intracranial pressure increases - the patient complains of a constant headache.
NB!
The intensity of manifestation of pituitary craniopharyngioma depends not only on the size and location of the tumor. If the tumor is large, but it does not compress neighboring areas, the symptoms of the pathology may be minor.
Locations and types of craniopharyngiomas
Depending on the location, several types of craniopharyngiomas are distinguished. Below is a detailed topographic classification:
- First type. Neoplasia is localized in the anterior part of the pituitary stalk. There are three subtypes:
- A – the tumor is formed under the diaphragm of the sella turcica (DS);
- B – in the anterior part of the optic chiasm and above the DTS;
- C – among the pituitary stalk and the optic chiasm (above the DTS).
- Second type. It includes tumors that form immediately above the pituitary stalk. As a rule, in this case, pressure is exerted on the third ventricle of the brain.
- Third type. Craniopharyngioma is located behind the pituitary stalk. In this case, three subtypes are distinguished:
- A – under the diaphragm of the sella turcica;
- B – under DTS. Often, neoplasia in this case reaches the anterior region of the pons;
- C – on the DTS, while reaching the cerebellopontine angle.
- Fourth type. The neoplasm is localized in the region of the pituitary stalk, usually in the area of the basal ganglia.
- Fifth type. The tumor is located in the 3rd and 4th ventricles of the brain, and in the area of the appendages of the nasal sinuses.
Note. The classification of craniopharyngiomas is of a directly applied nature. The choice of surgical approach directly depends on the type of tumor.
Diagnostics
To diagnose craniopharyngioma, several studies are performed at once. This helps determine the possible cause of the pathology, its exact location, and the degree of damage to surrounding tissues. Standard diagnosis of craniopharyngioma includes the following measures:
- Anamnesis collection, visual examination of the patient
. The doctor will learn about the symptoms that concern the person, clarify the general state of health, and the presence of risk factors; - Biochemical and general blood tests
. Determine the concentration of vital blood components; - General urine analysis
. Determines kidney function, rate of urine excretion; - Hormonal blood test
. Determines the content of biochemically active substances and identifies deviations from the norm. The test allows you to diagnose endocrine disorders; - Neurological examination
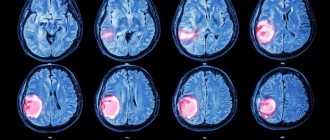
. The function of the brain, spinal cord, and nerve endings is assessed. The doctor needs to check the person’s muscle strength, memory, vision, hearing, reflexes, and coordination; - CT and MRI
. Diagnostic imaging techniques that provide accurate images of the brain. This helps determine the size and location of the tumor and what tissues it has affected. MRI when determining pituitary craniopharyngioma gives a clearer picture; - Biopsy
. The most accurate examination method for craniopharyngioma. A portion of the tumor is removed using a needle for further histological examination. This determines the exact nature of the tissues.
NB!
When diagnosing pituitary craniopharyngioma, blood and urine tests are of high diagnostic value. Your doctor will need to check your electrolyte levels to help diagnose diabetes insipidus. It is also necessary to exclude germ cell tumors, for which the levels of alpha-fetoprotein (α-FP) and beta human chorionic gonadotropin (β-hCG) are determined.
3.Diagnosis of the disease
Diagnosis of tumors is carried out using magnetic resonance imaging (MRI) and computed tomography (CT)
.
In addition, hormonal studies
and plain
radiography of the skull
. A set of studies allows us to determine the exact location and size of the tumor, its structure and the degree of infiltration into the tissue of neighboring brain structures.
About our clinic Chistye Prudy metro station Medintercom page!
Treatment
The basic treatment for pituitary craniopharyngioma is chemotherapy. It is carried out both as the main part of therapy and as an auxiliary part during surgical excision of the tumor. The radioactive drug is injected intravenously or into the spinal canal (in the case of surgical removal of a tumor, in its bed). Craniopharyngiomas cysts are filled with interferon preparations: they have a cytotoxic effect. Due to this, the process of self-destruction of tumor cells is started.
Craniopharyngiomas are prone to recurrence. And even after surgical interventions, many patients may develop a new formation within the first year. Because of this, they always undergo postoperative radiation therapy, which reduces the risk of such a consequence by 2 times. Today, the following methods show the greatest effectiveness in the treatment of pituitary craniopharyngiomas:
- Proton therapy
. Heavily charged particles are used that release all the energy into the tumor. Due to such targeted irradiation, it is possible to stop the growth of the tumor and also destroy some of the cells. The advantage of this technique is its highly precise action: the surrounding brain tissue is not irradiated; - Intracavitary radiotherapy
. This is one of the methods of radiotherapy in which the radiation source is located directly in the tumor. This avoids damage to the cranial nerves, blood vessels, pituitary gland and hypothalamus.
Therapy for pituitary craniopharyngioma in children
If pituitary craniopharyngioma is detected in children under 3 years of age, and the tumor is accompanied by the spread of numerous cysts, then a catheter is implanted in the pituitary gland area. Drainage of the cyst is organized, due to which its size is reduced. Nearby tissues and blood vessels are no longer compressed, and the symptoms of the pathology disappear.
Cyst catheterization is a minimally invasive method for treating pituitary craniopharyngioma. To increase the accuracy of the intervention, stereotactic settings are used. To remove cysts, a sclerosing agent may be injected into them. The walls of the tumor stick together, the tumor stops accumulating fluid. This technique is used exclusively in young children; in the future, more radical intervention is necessary: radiation therapy or surgical excision.
In older children, craniopharyngiomas are removed with a gamma knife. This is one of the methods of stereotactic radiosurgery, characterized by extreme targeting accuracy. High-dose radiation therapy is performed to remove the tumor. In this case, the surrounding tissues are not affected.
Types of operations to remove craniopharyngioma
Surgical treatment is the most effective method of treating cerebral craniopharyngioma
. The operation is quite complex: it has technical difficulties due to limited accessibility to the tumor. There is also a risk of damage to the pituitary gland, optic nerves, carotid arteries, and the floor of the third ventricle - all of these tissues are located in close proximity. The surgeon needs to remove all tumor tissue and not affect healthy cavities.
Radical tumor removal reduces the risk of relapse by up to 20%. In the future, postoperative radiation therapy can minimize the likelihood of future tumors. Currently, the following surgical techniques are used for pituitary craniopharyngioma:
- Surgery with craniotomy access with traditional opening of the cranial bones
. All manipulations are determined in advance using computer planning. This allows the operation to be performed as carefully as possible, preserving all surrounding tissues and formations; - Operation with transsphenoidal approach
. This is an endoscopic type of intervention. Small incisions are made under the nose and sinuses of the sphenoid bones, through which a neuroendoscope is inserted into the brain cavity. This minimally invasive intervention is easier for patients to tolerate and the rehabilitation period is shortened. But it must be taken into account that not all craniopharyngiomas are suitable for such an operation. Interventions with a transsphenoidal approach are performed only when the tumor is located suprasellar.
Forecast
The prognosis depends on the stage and nature of the tumor, the correctness of treatment and the individual characteristics of the patient. Mortality after tumor removal is no higher than 5-10%, and this number includes mortality for other reasons that are not directly related to the tumor process, so we can talk about lower mortality. Five-year survival rate – 50 – 87%. As a rule, most relapses develop within a year after surgery, a smaller part within three years. Recurrence has a 7-33% chance of occurring, usually occurring within the first five years after tumor removal.
Postoperative changes after removal of craniopharyngioma
Almost all patients with craniopharyngioma experience complications that impair quality of life. Such consequences can arise either due to the tumor itself or as a result of treatment. Patients often encounter the following problems:
- Loss of vision;
- The appearance of mood swings;
- Emotional disorders;
- Memory impairment;
- Endocrine dysfunction;
- Development of metabolic syndrome;
- Pathologies of blood vessels.
The pituitary gland is the main gland in the human body. It is she who is responsible for the production of basic hormones, and also controls the work of other organs and endocrine glands. If a person's pituitary gland function is impaired, multiple problems arise: obesity, decreased bone mineral density, deterioration of the lipid profile, decreased fertility. This often occurs even in those patients who adhere to hormone replacement therapy.
Due to damage to the hypothalamus, multiple hormonal changes also occur. And this affects the quality of life. The patient begins to complain of increased appetite, his body weight rapidly increases, his behavior changes, and his personality becomes emotional. Metabolic syndrome often develops - a group of disorders accompanied by obesity, elevated levels of triglycerides and cholesterol, and high blood pressure. Metabolic syndrome significantly increases the likelihood of strokes and heart attacks.
In most cases, treatment for pituitary craniopharyngioma requires surgery or radiation therapy. After such events, the likelihood of disorders of the blood vessels of the brain increases significantly. The risk of aneurysms and stroke increases, seizures and cognitive impairment often occur.
Due to the high risk of complications associated with pituitary craniopharyngioma, the patient must be under constant medical supervision. Only timely diagnosis will make it possible to identify the deviation at the initial stages and stop it. Social and psychological assistance will help improve the patient's condition. The specialist will tell the person how to competently cope with the experience, how to get rid of anxiety and minimize the risk of depression.
4. Treatment of craniopharyngioma
Treatment of craniopharyngioma
usually done through surgery. The purpose of the operation is radical excision of the tumor, which eliminates compression of the pituitary gland and normalizes its function in producing hormones.
For suprasellar localization of craniopharyngioma, surgery is usually performed via a transcranial approach using craniotomy. However, if the size and location of the tumor allow, it is possible to perform a more gentle operation using the transsphenoidal endoscopic method. This type of surgery is performed using innovative endoscopic equipment through the nasal passages.
. This intervention is less traumatic for the patient, it allows him to recover in a shorter time and reduces the risk of possible postoperative complications.
If the tumor is difficult to reach or surgery is contraindicated for the patient, treatment with stereotactic radiation therapy
, allowing extremely precise targeting of the beam gun to the tumor area without damaging adjacent tissues.
Prognosis and prevention
At the moment, doctors have not developed an unambiguous system for determining the stage of craniopharyngioma. The tumor can be newly diagnosed or recurrent. This is a benign neoplasm that is not prone to spreading to other parts of the brain. Despite this, they can cause numerous abnormalities in the body’s functioning: disruption of endocrine functions, damage to the optic nerve.
The 10-year survival rate for patients with pituitary craniopharyngioma is 90%. With adequate treatment, it is possible to preserve both the quality and life expectancy of the patient. If after radiation therapy the tumor continues to progress, they are completely removed.
It is impossible to completely protect yourself from pituitary craniopharyngioma. The disease has a hereditary predisposition. The only way to reduce the likelihood of developing pathology is to prevent the exacerbation of diseases in the mother during pregnancy, avoid self-medication with medications, and protect against the influence of toxins and poisons.
Diagnosis of craniopharyngioma
Diagnosis of craniopharyngioma begins with a consultation with a neurologist and oncologist. Next, an X-ray of the brain is prescribed, which reveals the structure and erosion of its walls characteristic of the tumor, as well as corresponding changes in the structure of the sella turcica. Calcified tumors are clearly identified by such studies.
Laboratory tests are also performed to determine the concentration of hormones in the blood.
The tumor is diagnosed using MRI and CT scans of the pituitary gland of the brain, which allow one to see the layers of brain structures and determine not only the location of the craniopharyngioma, but also its size, which is extremely important.
Sometimes Dopplerography of cerebral vessels, as well as angiography of the brain and spinal cord, are prescribed.
To make an adequate diagnosis, craniopharyngioma should be differentiated from a number of other tumors with similar symptoms, for example, pituitary adenoma, glioma, colloid cyst of the third ventricle.
conclusions
Craniopharyngioma is a benign neoplasm of the pituitary gland. Mostly occurs in children 5-14 years old. The disease develops due to disorders that occur in the fetus in the first trimester of pregnancy. In the initial stages, it is difficult to diagnose, because the pathology does not manifest itself with any symptoms for a long time. Treatment for pituitary craniopharyngioma is mainly surgical.
If you suspect pituitary craniopharyngioma in yourself or cannot recover from this disease, then feel free to contact the doctors of our medical center. They will conduct extensive diagnostics, develop an individual treatment regimen and monitor all stages of therapy.
Histological picture
1. Adamantinoma-like (about 85% of all craniopharyngiomas) - more common in patients of childhood and adolescence. They differ in polymorphism and can have a compact (10%), cystic (30%) and mixed (60%) structure.
2. Papillomatous (15% of all craniopharyngiomas) - unlike adamantine-like ones, this pathology is diagnosed more often in people in adulthood, usually at 30-40 years of age. These tumors are mostly solid, with little or no petrification